Causes of the disease
Short neck syndrome, in fact, is not a disease, but a special anomaly in the development of the spine, leading to the occurrence of many other diseases of the spine.
The development of this disease in the prenatal period can occur as a result of:
- segmentation;
- vascularization disorders; aplasia;
- delayed fusion in the fetal and embryonic periods of paired formation of vertebrae;
- hypoplasia.
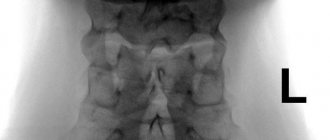
Formed synostoses of the upper thoracic and cervical vertebrae, a decrease in the number of cervical vertebrae to 4 - 5, non-fusion of the vertebral bodies and arches allow us to ultimately determine not only the general clinical picture, but also to understand how severe the deformation is in this syndrome.
All experts identify hereditary factors that contribute to the occurrence of Klippel-Feil syndrome:
- Genetic hereditary defect in chromosome 12 or 5, 8. In a sick child, there is a complete disruption of the formation of the so-called growth differentiation, which is necessary for the normal further development of the skeleton (including the formation of boundaries between joints and bones). This inevitably leads over time to disruption of the formation and formation of the upper thoracic and cervical vertebrae in the 3rd - 8th week of development inside the womb.
- Autosomal dominant type of inheritance of the disease. With this form of inheritance in a family in which one of the parents is sick, the probability of having a sick child is from 50 to 100 percent. This type of inheritance occurs most often in Klippel-Feil syndrome.
- Autosomal recessive type of inheritance of the disease. In this case, in a family in which one of the parents is sick, the probability of having a sick child is from zero to 50 percent.
Symptoms and course of the disease
The main signs of short neck syndrome are:
- low back hairline;
- shortened neck (brevicollis);
- “proud head position” (the head tilts slightly backward);
- restriction of neck mobility - may not be noticeable if there is fusion of fewer than three vertebrae or if fusion is limited to only the lower cervical vertebrae. Mobility limitations are more noticeable with rotation than with flexion/extension or lateral flexion of the neck.
The following symptoms may also occur, the presence of which is not mandatory, but with Klippel-Feil syndrome they occur quite often:
- scoliosis - sideways curvature of the spine;
- facial asymmetry;
- torticollis (curvature of the neck);
- wrinkling of the skin of the neck;
- high shoulder blade;
- weakness of facial muscles (paralysis);
- cleft palate;
- deafness.
It is important to note that Klippel-Feil syndrome is quite often combined with many other anomalies. So, in approximately 25 - 30 percent of patients the following are determined upon examination:
- scoliosis;
- bone rigid form of torticollis;
- wing-shaped folds on the neck;
- high position of the shoulder blades - Sprengel's disease.
In addition, Klippel-Feil syndrome may be accompanied by the development of abnormalities of the lower and upper extremities:
- hypoplasia of the first finger of the hand;
- syndactyly;
- extra fingers;
- absence of the ulna;
- hypoplasia of the pectoral muscle;
- hypoplasia of the upper limb;
deformation of the feet. Hidden anomalies in the development of internal organs in patients with this disease pose a greater danger to their lives than Klippel-Feil syndrome itself. The fact is that approximately 35 percent of patients have very severe and dangerous anomalies of kidney development in the form of hypoplasia of the pelvis, aplasia, hypoplasia, ectopia of the ureters and hydronephrosis. In addition, anomalies in the development of the cardiac and vascular systems are quite often observed (patent fusion of the ductus arteriosus, defects in the development of the interventricular septum, absence of a lung, dextroposition of the aorta, and others).
Disruption of the development of the entire spinal column is often accompanied by serious disruptions in the development of the child’s nervous system: at an early age, negative changes in the central nervous system can manifest themselves in the form of synkinesis, that is, friendly, involuntary movements of the arms and hands; at an older age, neurological symptoms in most cases are supplemented by complex secondary changes in the roots and spinal cord, which developed as a result of various degenerative changes in the spine, as well as due to protrusion of the cranial base and narrowing of the entire spinal canal.
Forms
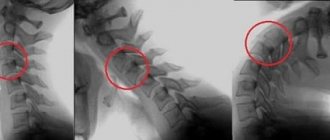
Based on X-ray data, the following forms of Klippel-Feil syndrome are distinguished:
- fusion of several cervical vertebrae;
- reduction in the size of the cervical vertebrae;
- absence of several cervical vertebrae.
A combination of these signs is possible.
Treatment of the disease
Many people believe that this syndrome is incurable. However, in order to understand whether this disease can be cured and whether surgery will help, you should undergo examination by a highly qualified specialist.
If you have Klippel-Feil syndrome, you should contact a traumatologist and surgeon. You may also need to consult a medical geneticist and neurosurgeon.
- Non-surgical methods (ineffective):
- a set of exercises, physical therapy and massage that increase the mobility of the cervical spine;
- fixation of the cervical spine with a special collar;
- Drug therapy is resorted to in those severe cases when the patient experiences compression of the roots (nerves) and severe pain. In this situation, doctors rarely prescribe anti-inflammatory non-steroidal drugs, since they are ineffective. And since inflammation is not observed in patients with Klippel-Feil syndrome, doctors prescribe them traditional analgesics and prescribe various physiotherapeutic procedures that are designed to reduce pain.
- Surgical methods:
- to increase the mobility of the neck, 3-4 upper ribs are removed (cervicalization according to Bonola) - the surgeon makes a paravertebral incision along the edge of the scapula (inner) and the spinous processes. From the edge of the scapula, the doctor cuts off the rhomboid and trapezius muscles - they are retracted inward, and the scapula outward. Then, over a distance of 10-18 cm, the doctor resects the 1st-4th ribs and removes the periosteum. Afterwards, immobilization is carried out with a plaster bed, and later with a polyethylene head holder. The operation is performed on one side, and when the recovery period is complete, the operation is performed on the other side. After the operation, the prognosis is favorable, the neck not only becomes longer, but also acquires significantly greater mobility;
- fixation of underdeveloped vertebrae with special metal pins.
How is an appointment with a vertebroneurologist?
There is no need to specially prepare your child for visiting the clinic. During the consultation, the doctor will listen to complaints and ask about the presence of clinical symptoms. Information about past diseases and surgical interventions will help the doctor compile an anamnesis.
During the consultation, considerable importance is given to a physical examination of the child, during which the specialist visually assesses the general condition of the patient and decides on the advisability of additional diagnostics.
Diagnosis of the disease
Diagnosis of Klippel-Feil-Sprengel syndrome is based on a combination of symptoms: shortening of the neck observed from birth, low hair growth on the neck and limited head mobility.
To clarify the type of deformation, an X-ray examination of the cervical and thoracic spine is performed. X-ray of the cervical and upper thoracic spine (spondylography) allows you to accurately assess the condition of the vertebrae, identify their reduction and fusion. X-ray examination is also carried out in the position of maximum flexion and extension of the neck to identify instability in the cervical spine. The images often reveal the fusion of 4-6 vertebrae into a solid bone mass.
Ultrasonography:
- heart (often Klippel-Feil syndrome is combined with patent interventricular septum, patent Botallian duct, which closes 1-2 weeks after birth);
- kidneys (sometimes with Klippel-Feil syndrome one kidney is missing).
ECG (electrocardiography) can detect signs of cardiac dysfunction.
Research of the family tree - through a detailed conversation with the patient and his relatives. Information about the presence of similar complaints in relatives, as well as the age of development of the first symptoms and the degree of relationship (close, distant) is important.
Genetic research is carried out on the patient and family members in order to search for a genetic defect that is inherited.
It is also possible to consult a neurosurgeon or medical geneticist.
Differential diagnosis is carried out with tuberculous spondylitis of the upper cervical vertebrae, bilateral and unilateral forms of muscular and other types of torticollis (especially in the absence of effect from conservative treatment).
Basic indications for visiting a specialist
You should not postpone a visit to a pediatric vertebrologist if you find the following signs of spinal column pathology in your son or daughter:
- The slightest signs of curvature of the spine (as a rule, this occurs at 6-8 years);
- Folds, depressions, birthmarks on the back may be signs of hidden spina bifida, which also requires immediate medical correction;
- Congenital defect of the skin on the back (manifests itself in the form of a hole in which nerves and fragments of the spinal cord can be seen);
- Frequent dizziness, complaints of headache;
- State of chronic fatigue, fatigue, feeling of general weakness;
- Numbness, tingling, sensation of “pins and needles” in the limbs;
- Disturbances in the functioning of the vestibular apparatus.
Prices
| Disease | Approximate price, $ |
| Prices for hip replacement | 23 100 |
| Prices for clubfoot treatment | 25 300 |
| Prices for Hallux Valgus treatment | 7 980 |
| Prices for knee joint restoration | 13 580 — 27 710 |
| Prices for scoliosis treatment | 9 190 — 66 910 |
| Prices for knee replacement | 28 200 |
| Prices for treatment of intervertebral hernia | 35 320 — 47 370 |
What tests may be required to make a diagnosis?
Depending on how much information the vertebroneurologist initially has, he can refer the child to undergo the missing tests and examinations. So, the list of diagnostic procedures may include:
- Laboratory blood tests;
- Ultrasound of different parts of the spine;
- Computed tomography (the method allows you to exclude or confirm the presence of an oncological tumor that puts pressure on the nerve);
- X-ray of the spine;
- MRI;
- X-ray of the spine using a contrast agent.
You can undergo many of these and other manipulations in Kaliningrad in the diagnostic department of our MedProfi clinic. The final choice of diagnostic procedures is made by the doctor on an individual basis.
Diagnosis of Shereshevsky–Turner syndrome
General diagnostic criteria for all forms of Shereshevsky-Turner syndrome are height less than 150 cm, primary amenorrhea, underdevelopment of the genital organs, absence or poor development of secondary sexual characteristics, according to ultrasound examination - a reduced uterus with a linear endometrium, and ovaries without follicles. Blood tests reveal elevated levels of gonadotropins, particularly follicle-stimulating hormone, and decreased levels of estrogen. Karyotyping allows you to identify a defective set of chromosomes, with the absence or significant reduction of sex chromatin.
Differential diagnosis of Shereshevsky-Turner syndrome is carried out with primary amenorrhea of hypothalamic origin. Unlike the latter, patients with Shereshevsky-Turner syndrome do not have neuropsychiatric symptoms.
Treatment of Shereshevsky–Turner syndrome
The choice of treatment tactics for patients with Shereshevsky-Turner syndrome depends on the presence or absence of the Y chromosome in the karyotype. If elements of the Y chromosome are detected in the karyotype, the ovaries are surgically removed at a young age (up to 20 years), in order to prevent the degeneration of the gland tissue into a malignant tumor.
In cases where there is no Y chromosome in the karyotype of patients, or after surgical removal of the ovaries, hormone replacement therapy with estrogen is prescribed at the age of 16-18 years, the purpose of which is the development of secondary sexual characteristics (female hair growth, enlarged mammary glands), reducing the level of gonadotropins, restoring menstrual cycle, preventing the development of estrogen deficiency conditions - osteoporosis, metabolic disorders, diseases of the cardiovascular system, improving the quality of life and adaptability to society. If necessary, patients are provided with psychological assistance.
Short stature is corrected by administering growth hormone until the growth plates are closed. The prognosis of Shereshevsky-Turner syndrome is favorable in terms of life expectancy and intellectual development. With regard to restoration of reproductive function, the prognosis is unfavorable; most patients remain infertile.
Shereshevsky-Turner syndrome - signs of the disease
Patients with Shereshevsky-Turner syndrome complain of lack of menstrual flow, short stature and some cosmetic defects in appearance.
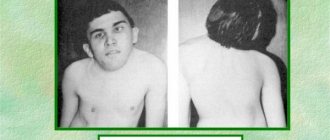
Most newborn girls with this disease have only mild clinical manifestations, but a certain group of infants have lymphedema of the hands and feet, as well as skin folds on the back of the neck. Some of the most common anomalies found in Shereshevsky-Turner disease are a short neck with low hair growth and wing-shaped cervical folds, an expanded chest, poorly developed genitals - the labia minora, clitoris, uterus, as well as widely spaced inverted nipples. Some patients have abnormalities in the shape and location of the ears (protruding ears), which are combined in half of the cases with hearing loss.
Compared to their relatives, girls are shorter - no more than 150 cm, but at the same time their body weight is quite impressive. Less commonly, signs such as multiple pigmented nevi or vitiligo, anomalies of the metacarpal and metatarsal bones of the hands and feet, deformation of the elbow and shoulder joints, and hypoplastic narrow nails are detected. The facial part of the patients is also modified - the jaw bones are reduced in size, the palate is high, and drooping eyelids are also observed.
From the internal organs, it is possible to detect cardiac anomalies in the form of coarctation of the aorta, its dissection, disruption of the integrity of the interventricular septum and bicuspid aortic valve. Congenital anomalies and malformations of the kidneys (horseshoe kidney, duplication of the pelvis and ureters), and blood vessels of a tumor nature (hemangiomas, telangiectasia) are also common.
The female gonads are replaced by strands of connective tissue that are unable to produce germ cells, as a result of which the vast majority (90%) of sick girls do not go through puberty, the mammary glands do not enlarge, and primary amenorrhea develops. In the remaining 10%, the spontaneous onset of menstruation is possible, but even in this case, the woman’s fertility remains in question.
The mental sphere of patients with Shereshevsky-Turner syndrome practically does not suffer. True, some authors note a decrease in their attention and some processes of perception, in particular spatial, which affects the quality of processing of nonverbal (nonverbal) information.
There are three forms of gonadal dysgenesis - erased, pure and mixed, which differ from each other in clinical manifestations. When the form is erased
In patients, a mosaic karyotype of 45 X0/46 XX is detected, and the severity of the clinical picture is associated with the ratio of cells with a normal and deformed karyotype. If the percentage of cells with the absence of one of the X chromosomes predominates, then the external signs of the patient will be similar to the classic appearance of patients with Shereshevsky-Turner disease. Otherwise, spontaneous development of external sexual characteristics will be noted, but patients will have primary amenorrhea and signs of underdevelopment of the genital organs.
Pure form of ovarian dysgenesis (Swyer syndrome)
characterized by karyotype 46 XX or 46 XY. It is difficult to recognize the presence of chromosomal defects by appearance, since the growth of patients is normal, somatic defects and anomalies are not detected. However, external sexual characteristics are poorly developed. In addition, upon examination, genital infantilism is revealed, i.e. the genitals are also underdeveloped.
Mixed form of ovarian dysgenesis
manifested by primary amenorrhea, virilization - enlargement of the clitoris, male-type hair growth, which is due to the presence of an inferior Y-chromosome in the karyotype. With this form of the disease in the postpubertal period, combined type ovarian tumors are possible - gonadoblastomas, embryonal carcinomas.