Причины заболевания
Синдром короткой шеи, по сути, не болезнь, а особая аномалия развития позвоночника, приводящая к возникновению многих других болезней позвоночника.
Развитие данного заболевания в внутриутробный период может произойти в результате:
- сегментации;
- нарушения васкуляризации; аплазии;
- задержки слияния в фетальном и эмбриональном периодах парного формирования позвонков;
- гипоплазии.
Сформировавшиеся синостозы верхних грудных и шейных позвонков, уменьшение количества шейных позвонков до 4х — 5-ти, несращение тел и дуг позвонков позволяют по итогу определить не только общую клиническую картину, но и понять, насколько сильна деформация при этом синдроме.
Все специалисты выделяют наследственные факторы, которые способствуют возникновению синдрома Клиппеля-Фейля:
- Генетический наследственный дефект в 12 или 5, 8 хромосоме. У больного ребенка при этом полностью наблюдается нарушение образования так называемой дифференциации роста, необходимой для нормального дальнейшего развития скелета (в том числе, для образования между суставами и костями границ). Это неизбежно приводит со временем к нарушению формирования и закладки верхних грудных и шейных позвонков на 3-ей — 8-ой неделе развития внутри утробы матери.
- Аутосомно-доминантный тип наследования болезни. При этой форме наследования в семье, в которой один из родителей болеет, вероятность появления на свет больного ребенка составляет от 50-ти до 100 процентов. Данный тип наследования встречается чаще всего при синдроме Клиппеля-Фейля.
- Аутосомно-рецессивный тип наследования заболевания. В этом случае в семье, в которой один из родителей болеет, вероятность появления на свет больного ребенка составляет от нуля до 50-ти процентов.
Симптомы и протекание заболевания
Основными признаками синдроа короткой шеи являются:
- низкая задняя линия роста волос;
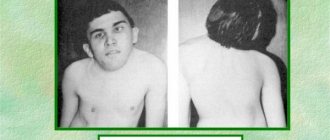
- укороченная шея (бревиколлис);
- «гордая посадка головы» (голова слегка отклоняется кзади);
- ограничение подвижности шеи — может быть незаметным, если имеется сращение менее трех позвонков или если сращение ограничено только нижними шейными позвонками. Ограничения подвижности более заметны при вращении, чем при сгибании/разгибании или боковом сгибании шеи.
Также могут встречаться следующие признаки, наличие которых не является обязательным, но при синдроме Клиппеля-Фейля они встречаются довольно часто:
- сколиоз — искривление позвоночника вбок;
- асимметрия лица;
- кривошея (искривление шеи);
- сморщивание кожи шеи;
- высоко расположенная лопатка;
- слабость мышц лица (параличи);
- расщепления неба;
- глухота.
Важно отметить, что синдром Клиппеля-Фейля довольно часто сочетается со многими другими аномалиями. Так, примерно у 25-ти — 30-ти процентов больных определяются при осмотре:
- сколиоз;
- костная ригидная форма кривошеи;
- крыловидные складки на шее;
- высокое расположение лопаток — болезнь Шпренгеля.
Кроме того, синдром Клиппеля-Фейля может сопровождаться развитием аномалии нижних и верхних конечностей:
- гипоплазией первого пальца кисти;
- синдактилией;
- дополнительными пальцами;
- отсутствием локтевой кости;
- гипоплазией грудинной мышцы;
- гипоплазией верхней конечности;
деформацией стоп. Скрытые аномалии в развитии внутренних органов у пациентов с этим заболеванием собой представляют большую опасность для их жизни, чем сам синдром Клиппеля-Фейля. Дело в том, что примерно у 35-ти процентов больных имеются весьма тяжелые и опасные аномалии развития почек в форме гипоплазии лоханок, аплазии, гипоплазии, эктопии мочеточников и гидронефроза. Помимо этого, довольно часто наблюдаются аномалии развития сердечной и сосудистой систем (незаращение боталлова (артериального) протока, дефекты в развитии межжелудочковой перегородки, отсутствие легкого, декстропозиция аорты и другие).
Нарушение развития всего позвоночного столба нередко сопровождается серьезными сбоями в развитии у ребенка его нервной системы: в раннем возрасте негативные изменения со стороны центральной нервной системы могут проявляться в форме синкинезий, то есть, содружественных, непроизвольных движениях рук, кистей; в более старшем возрасте неврологическая симптоматика в большинстве случаев дополняется сложными вторичными изменениями корешков и спинного мозга, развившимися в результате дегенеративных различных изменений позвоночника, а также из-за протрузии черепного основания и сужения всего позвоночного канала.
Формы
На основе данных рентгенологического исследования выделяют следующие формы синдрома Клиппеля-Фейля:
- сращение нескольких шейных позвонков;
- уменьшение размеров шейных позвонков;
- отсутствие нескольких шейных позвонков.
Возможна комбинация названных признаков.
Лечение заболевания
Многие люди считают, что данный синдром неизлечим. Однако для того, чтобы понять, можно ли вылечить данное заболевание и поможет ли операция, следует пройти обследований у высококвалифицированного специалиста.
С синдромом Клиппеля-Фейля следует обращаться к травматологу и хирургу. Также, возможно, понадобится консультация медицинского генетика и нейрохирурга.
- Нехирургические методы (малоэффективны):
- комплекс упражнений, лечебная физкультура и массаж, увеличивающие подвижность шейного отдела позвоночника;
- фиксация шейного отдела позвоночника специальным воротником;
- к медикаментозной терапии обращаются уже в тех тяжелых случаях, когда у больного появляются компрессия корешков (нервных) и сильные болевые ощущения. В этой ситуации доктора редко назначают противовоспалительные нестероидные препараты, поскольку они малоэффективны. И так как воспаления у больных синдромом Клиппеля-Фейля не наблюдается, врачи прописывают им традиционные анальгетики и назначают различные физиотерапевтические процедуры, которые призваны понижать болевой синдром.
- Хирургические методы:
- для увеличения подвижности шеи удаляют 3-4 верхних ребра (цервикализация по Бонола) — хирург производит паравертебральный разрез по меж краем лопатки (внутренним) и остистыми отростками. От края лопатки врач отсекает ромбовидную и трапециевидную мышцы — их отводят внутрь, а лопатку кнаружи. Затем, на протяжении 10-ти — 18-ти см доктор резецирует I-IV ребра и удаляет надкостницу. После осуществляют иммобилизацию гипсовой кроваткой, а позже — полиэтиленовым головодержателем. Операция выполняется на одной стороне, а когда завершается период восстановления, операция проводится на другой стороне. После операции прогноз благоприятный, шея не только становится длиннее, но и приобретает значительно большую подвижность;
- фиксация недоразвитых позвонков специальными металлическими штифтами.
Как проходит прием вертеброневролога?
Специально готовить ребенка к посещению клиники нет необходимости. Во время консультации врач выслушает жалобы и расспросит о наличии клинических симптомов. Информация о перенесенных заболеваниях и хирургических вмешательствах поможет доктору составить анамнез.
Немаловажное значение во время консультации уделяется физикальному осмотру ребенка, в ходе которого специалист визуально оценивает общее состояние пациента, принимает решение о целесообразности проведения дополнительной диагностики.
Диагностика заболевания
Диагностика синдрома Клиппеля-Фейля-Шпренгеля основана на сочетании симптомов: укорочение шеи, наблюдаемое с рождения, низкая граница роста волос на шее и ограничение подвижности головы.
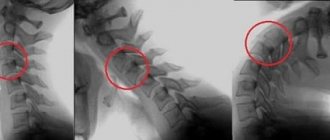
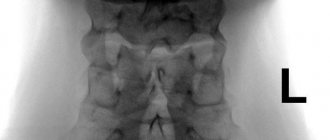
Для уточнения типа деформации проводят рентгенологическое исследование шейного и грудного отделов позвоночника. Рентгенография шейного и верхнегрудного отделов позвоночника (спондилография) позволяет точно оценить состояние позвонков, выявить их уменьшение, сращение. Также ренгтенологическое исследование проводится в положении максимальных сгибания и разгибания шеи, чтобы выявить нестабильность в шейном отделе позвоночника. На снимках чаще выявляют сращение 4-6 позвонков в сплошную костную массу.
Ультразвуковое исследование:
- сердца (нередко синдром Клиппеля-Фейля сочетается с незаращением межжелудочковой перегородки, незаращением Боталлова протока, который закрывается на 1-2 неделе после рождения);
- почек (иногда при синдроме Клиппеля-Фейля отсутствует одна почка).
ЭКГ (электрокардиография) позволяет обнаружить признаки нарушения работы сердца.
Исследование генеалогического дерева — с помощью подробной беседы с больным и его родственниками. Имеет значение информация о наличии подобных жалоб у родственников, а также возраст развития первых симптомов и степень родства (близкое, далекое).
Генетическое исследование проводится у больного и у членов семьи с целью поиска генетического дефекта, передающегося по наследству.
Возможна также консультация нейрохирурга, медицинского генетика.
Дифференциальный диагноз проводят с туберкулезным спондилитом верхних шейных позвонков, двусторонней и односторонней формами мышечной и иными типами кривошеи (особенно при отсутствии эффекта от консервативного лечения).
Основные показания для посещения специалиста
Не стоит откладывать визит к детскому вертебрологу при обнаружении у сына или дочери следующих признаков патологии позвоночного столба:
- Малейшие признаки искривления позвоночника (как правило, это происходит в 6-8 лет);
- Складки, углубления, родимые пятка на спине могут являться признаками скрытого расщепления позвоночника, что также требует незамедлительной медицинской коррекции;
- Врожденный дефект кожных покровов на спине (проявляется в виде отверстия, в котором можно рассмотреть нервы, фрагменты спинного мозга);
- Частые головокружения, жалобы на головную боль;
- Состояние хронической усталости, утомляемость, чувство общей слабости;
- Онемение, покалывание, ощущение «мурашек» в конечностях;
- Нарушения в работе вестибулярного аппарата.
Цены
| Заболевание | Ориентировочная цена, $ |
| Цены на протезирование тазобедренного сустава | 23 100 |
| Цены на лечение косолапости | 25 300 |
| Цены на лечение Халлюкс Вальгуса | 7 980 |
| Цены на реставрацию коленного сустава | 13 580 — 27 710 |
| Цены на лечение сколиоза | 9 190 — 66 910 |
| Цены на эндопротезирование коленного сустава | 28 200 |
| Цены на лечение межпозвоночной грыжи | 35 320 — 47 370 |
Какие исследования могут потребоваться для постановки диагноза?
В зависимости от того, каким объемом информации вертеброневролог располагает изначально, он может направить ребенка на прохождение недостающих анализов и обследований. Так, в перечень диагностических процедур могут войти:
- Лабораторные исследования крови;
- УЗИ разных отделов позвоночника;
- Компьютерная томография (метод позволяет исключить или подтвердить наличие онкологического новообразования, оказывающего давление на нерв);
- Рентгенография позвоночника;
- МРТ;
- Рентгеноскопия позвоночника с использованием контрастного вещества.
Многие из этих и других манипуляций в Калининграде вы можете пройти в диагностическом отделении нашей клиники «МедПрофи». Окончательный выбор диагностических процедур врач осуществляет в индивидуальном порядке.
Диагностика синдрома Шерешевского – Тернера
Общими диагностическими критериями для всех форм синдрома Шерешевского — Тернера служит рост менее 150 см, первичная аменорея, недоразвитие половых органов, отсутствие или слабое развитие вторичных половых признаков, по данным ультразвукового исследования — уменьшенная в размерах матка с линейным эндометрием, и яичники без фолликулов. При исследовании крови обнаруживается повышенный уровень гонадотропинов, в частности фоликулостимулирующего гормона, и пониженный уровень эстрогенов. Кариотипирование позволяет выявить дефектный набор хромосом, с отсутствием или существенным снижением полового хроматина.
Дифференциальная диагностика синдрома Шерешевского-Тернера проводится с первичной аменореей гипоталамического генеза. В отличии от последней у пациенток с синдромом Шерешевского — Тернера отсутствуют психоневрологические симптомы.
Лечение синдрома Шерешевского – Тернера
Выбор тактики лечения больных с синдромом Шерешевского — Тернера зависит от наличия или отсутствия в кариотипе У- хромосомы. Если в кариотипе обнаруживаются элементы Y-хромосомы, яичники удаляют оперативным путем в молодом возрасте (до 20 лет), с целью предотвращения перерождения тканей железы в злокачественную опухоль.
В случаях отсутствия Y-хромосомы в кариотипе пациенток, либо после хирургического удаления яичников назначают заместительную гормональную терапию эстрогенами в возрасте 16-18 лет, целью которой является развитие вторичных половых признаков (оволосение по женскому типу, увеличение молочных желез), снижение уровня гонадотропинов, восстановление менструального цикла, предупреждение развития эстрогендефицитных состояний — остеопороза, нарушений метаболизма, заболеваний сердечно-сосудистой системы, повышение качества жизни и адаптированность в социум. При необходимости больным оказывается психологическая помощь.
Низкорослость корригируется введением гормона роста до закрытия зон роста. Прогноз синдрома Шерешевского-Тернера благоприятный в отношении продолжительности жизни и интеллектуального развития. В отношении восстановления детородной функции прогноз неблагоприятный, большая часть пациенток остается бесплодной.
Синдром Шерешевского — Тернера — признаки заболевания
Больные с синдромом Шерешевского — Тернера предъявляют жалобы на отсутствие менструальных выделений, низкий рост и некоторые косметические дефекты внешности.
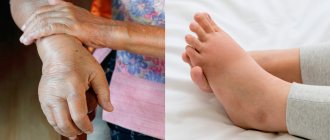
У большинства новорожденных девочек с этим заболеванием выявляются лишь легкие клинические проявления, однако у определенной группы младенцев отмечаются лимфатические отеки кистей и ступней, а также кожные складки на задней поверхности шеи. Одними из самых распространенных аномалий, встречающихся при заболевании Шерешевского — Тернера, считаются короткая шея с низким уровнем роста волос и крыловидными шейными складками, расширенная грудная клетка, слабо развитые половые органы — малые половые губы, клитор, матка, а также широко расставленные втянутые соски. У некоторых пациенток отмечаются аномалии формы и расположения ушей (лопоухость), которые сочетаются в половине случаев с тугоухостью.
В сравнении с родственниками девочки отличаются более низким ростом — не более 150 см, но при этом масса тела довольно внушительная. Реже выявляются такие признаки, как множественные пигментные невусы или витилиго, аномалии пястных и плюсневых костей кистей и стоп, деформация локтевых и плечевых суставов, гипоплазированные узкие ногти. Видоизменена и лицевая часть пациенток — челюстные кости уменьшены в размерах, небо высокое, наблюдается также опущение век.
Со стороны внутренних органов возможно обнаружение аномалий сердца в виде коарктации аорты, её расслоения, нарушения целостности межжелудочковой перегородки и двухстворчатого клапана аорты. Нередки и врожденные аномалии и пороки развития почек (подковообразная почка, удвоение лоханок и мочеточников), кровеносных сосудов опухолевой природы (гемангиомы, телеангиэктазии).
Женские половые железы замещаются тяжами из соединительной ткани, не способными вырабатывать половые клетки, в результате чего у подавляющего большинства (90%) больных девочек не наступает период полового созревания, молочные железы не увеличиваются, развивается первичная аменорея. У остальных 10% возможно спонтанное начало менструаций, но и в этом случае фертильность женщины остается под вопросом.
Психическая сфера больных с синдромом Шерешевского — Тернера практически не страдает. Правда, некоторые авторы отмечают снижение у них внимания и некоторых процессов восприятия, в частности пространственного, что отражается на качестве обработки невербальной (несловесной) информации.
Различают три формы дисгенезии гонад — стертая, чистая и смешанная, отличающиеся друг от друга клиническими проявлениями. При стертой форме
у пациенток выявляется мозаичный кариотип 45 Х0/ 46 ХХ, и выраженность клинической картины связана с соотношением клеток с нормальным и деформированным кариотипом. Если преобладает процент клеток с отсутствием одной из Х-хромосом, то внешние признаки больного будут схожи с классическим видом пациентов с заболеванием Шерешевского — Тернера. В противном случае будет отмечаться спонтанное развитие внешних половых признаков, но у больных будет выявлена первичная аменорея и признаки недоразвития половых органов.
Чистая форма яичниковой дисгенезии (синдром Свайера)
характеризуется кариотипом 46 ХХ, либо 46 ХY. По внешнему виду распознать наличие хромосомных дефектов сложно, так как рост пациенток нормальный, соматические дефекты и аномалии не выявляются. Однако, внешние половые признаки при этом слабо развиты. Кроме того, при осмотре выявляется генитальный инфантилизм, т.е. половые органы также недостаточно развиты.
Смешанная форма дисгенезии яичников
проявляется первичной аменореей, вирилизацией — увеличением клитора, оволосением по мужскому типу, что обусловлено наличием в кариотипе неполноценной Y-хромосомы. При этой форме заболевания в постпубертатном периоде возможны опухоли яичников комбинированного типа — гонадобластомы, эмбриональные карциномы.