Ibuprofen-Hemofarm tablet film 400 mg pack cont cell/pack card x30
Trade name: Ibuprofen-Hemofarm International name: Ibuprofen
Release form: film-coated tablets 400 mg (blisters)
Ingredients: ibuprofen 400 mg
Pharmacological group: NSAID (non-steroidal anti-inflammatory drug)
Pharmacological group according to ATK: M01AE01 (Ibuprofen)
Pharmacological action: antiplatelet, antipyretic, NSAID, anti-inflammatory, analgesic,
Indications: Inflammatory and degenerative diseases of the musculoskeletal system: rheumatoid, juvenile chronic, psoriatic arthritis, osteochondrosis, neuralgic amyotrophy (Personage-Turner disease), arthritis with SLE (as part of complex therapy), gouty arthritis (in case of an acute attack of gout, fast-acting medications are preferred forms), ankylosing spondylitis (ankylosing spondylosis). Pain syndrome: myalgia, arthralgia, ossalgia, arthritis, sciatica, migraine, headache (including menstrual syndrome) and toothache, cancer, neuralgia, tendinitis, tendovaginitis, bursitis, neuralgic amyotrophy (Personage-Turner disease) , post-traumatic and postoperative pain syndrome, accompanied by inflammation. Algodismenorrhea, inflammatory process in the pelvis, incl. adnexitis, childbirth (as an analgesic and tocolytic agent). Feverish syndrome with “colds” and infectious diseases.
Dosage regimen: Inside, after meals. Adults: for osteoarthritis, psoriatic arthritis and ankylosing spondylitis - 400-600 mg 3-4 times a day. For rheumatoid arthritis - 800 mg 3 times a day, for soft tissue injuries, sprains - 1.6-2.4 g / day in several doses. For algodismenorrhea - 400 mg 3-4 times a day, for moderate pain syndrome - 1.2 g / day. For children over 12 years of age, the initial dose is 150-300 mg 3 times a day, the maximum dose is 1 g, then 100 mg 3 times a day, for juvenile rheumatoid arthritis - 30-40 mg/kg/day in several doses. To reduce body temperature 39.2 degrees C and above - 10 mg/kg/day, below 39.2 degrees C - 5 mg/kg/day. Oral suspension - 5-10 mg/kg 3 times a day: children aged 6-12 months (only as prescribed by a doctor) - an average of 50 mg 3-4 times a day, 1-3 years - 100 mg 3 times per day, 4-6 years - 150 mg 3 times a day, 7-9 years - 200 mg 3 times a day, 10-12 years - 300 mg 3 times a day. For febrile syndrome after immunization - 50 mg, if necessary, after 6 hours, repeat administration at the same dose, maximum daily dose - 100 mg.
Contraindications: Hypersensitivity, erosive and ulcerative diseases of the gastrointestinal tract (including peptic ulcer of the stomach and duodenum in the acute stage, ulcerative colitis, peptic ulcer, Crohn's disease - nonspecific ulcerative colitis), "aspirin" asthma, blood clotting disorders (including hemophilia, prolongation of bleeding time, bleeding tendency, hemorrhagic diathesis), pregnancy, lactation.
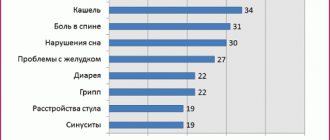
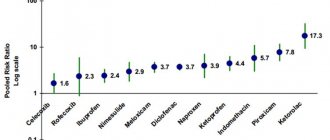
Side effects: From the digestive system: NSAID gastropathy (nausea, vomiting, abdominal pain, heartburn, loss of appetite, diarrhea, flatulence, pain and discomfort in the epigastric region), ulceration of the gastrointestinal mucosa (in some cases complicated by perforation and bleeding) , irritation, dryness of the oral mucosa or pain in the mouth, ulceration of the gum mucosa, aphthous stomatitis, pancreatitis, constipation, hepatitis. From the respiratory system: shortness of breath, bronchospasm. From the senses: hearing loss, ringing or noise in the ears, reversible toxic optic neuritis, blurred vision or diplopia, dryness and irritation of the eyes, swelling of the conjunctiva and eyelids (allergic origin), scotoma. From the nervous system: headache, dizziness, insomnia, anxiety, nervousness and irritability, psychomotor agitation, drowsiness, depression, confusion, hallucinations, rarely - aseptic meningitis (more often in patients with autoimmune diseases). From the cardiovascular system: development or worsening of heart failure, tachycardia, increased blood pressure. From the urinary system: acute renal failure, allergic nephritis, nephrotic syndrome (edema), polyuria, cystitis. Allergic reactions: skin rash (usually erythematous, urticaria), skin itching, angioedema, anaphylactoid reactions, anaphylactic shock, bronchospasm, fever, erythema multiforme exudative (including Stevens-Johnson syndrome), toxic epidermal necrolysis (Lyell's syndrome) , eosinophilia, allergic rhinitis. From the hematopoietic organs: anemia (including hemolytic, aplastic), thrombocytopenia and thrombocytopenic purpura, agranulocytosis, leukopenia. Other: increased sweating. The risk of developing ulcerations of the gastrointestinal mucosa, bleeding (gastrointestinal, gingival, uterine, hemorrhoidal), visual impairment (impaired color vision, scotoma, amblyopia) increases with long-term use in large doses. Overdose. Symptoms: abdominal pain, nausea, vomiting, lethargy, drowsiness, depression, headache, tinnitus, metabolic acidosis, coma, acute renal failure, decreased blood pressure, bradycardia, tachycardia, atrial fibrillation, respiratory arrest. Treatment: gastric lavage (only within an hour after administration), activated charcoal, alkaline drinking, forced diuresis, symptomatic therapy (correction of CBS, blood pressure).
Pharmacodynamics: NSAIDs have analgesic, antipyretic and anti-inflammatory effects due to non-selective blockade of COX1 and COX2 and have an inhibitory effect on Pg synthesis. The analgesic effect is most pronounced for inflammatory pain. Like all NSAIDs, ibuprofen exhibits antiplatelet activity.
Pharmacokinetics: Well absorbed from the gastrointestinal tract. Absorption is slightly reduced when taking the drug after meals. TCmax when taken on an empty stomach - 45 minutes, when taken after meals - 1.5-2.5 hours, in synovial fluid - 2-3 hours (where it creates higher concentrations than in plasma). 90% bound to plasma proteins. Subject to presystemic and postsystemic metabolism in the liver. After absorption, approximately 60% of the pharmacologically inactive R form of ibuprofen is slowly transformed into the active S form. The CYP2C9 isoenzyme takes part in the metabolism of the drug. It has two-phase elimination kinetics with T1/2 2-2.5 hours (for retard forms - up to 12 hours). It is excreted by the kidneys (no more than 1% unchanged) and, to a lesser extent, with bile.
Special instructions: During treatment, monitoring of the peripheral blood picture and the functional state of the liver and kidneys is necessary. When symptoms of gastropathy appear, careful monitoring is indicated, including esophagogastroduodenoscopy, a blood test to determine Hb, hematocrit, and a stool test for occult blood. To prevent the development of NSAID gastropathy, it is recommended to combine it with PgE drugs (misoprostol). If it is necessary to determine 17-ketosteroids, the drug should be discontinued 48 hours before the study. Patients should refrain from all activities that require increased attention, rapid mental and motor reactions. During the treatment period, ethanol intake is not recommended. Carefully. Cirrhosis of the liver with portal hypertension, hyperbilirubinemia, peptic ulcer of the stomach and duodenum (history), gastritis, enteritis, colitis, liver and/or renal failure, nephrotic syndrome, CHF, arterial hypertension, blood diseases of unknown etiology (leukopenia and anemia ), children's age (for tablet forms - up to 12 years, 6 months - for oral suspension). Children 6-12 months are prescribed only on the recommendation of a doctor.
Interaction: Inducers of microsomal oxidation (phenytoin, ethanol, barbiturates, rifampicin, phenylbutazone, tricyclic antidepressants) increase the production of hydroxylated active metabolites, increasing the risk of severe hepatotoxic reactions. Microsomal oxidation inhibitors reduce the risk of hepatotoxicity. Reduces the hypotensive activity of vasodilators (including BMCC and ACE inhibitors), natriuretic and diuretic activity - furosemide and hydrochlorothiazide. Reduces the effectiveness of uricosuric drugs, enhances the effect of indirect anticoagulants, antiplatelet agents, fibrinolytics (increasing the risk of hemorrhagic complications), ulcerogenic effect with bleeding of MCS and GCS, colchicine, estrogens, ethanol, enhances the effect of oral hypoglycemic drugs and insulin. Antacids and cholestyramine reduce the absorption of ibuprofen. Increases the blood concentration of digoxin, Li+ drugs and methotrexate. Caffeine enhances the analgesic effect. When administered simultaneously, ibuprofen reduces the anti-inflammatory and antiplatelet effects of ASA (an increase in the incidence of acute coronary insufficiency in patients receiving small doses of ASA as an antiplatelet agent is possible after starting ibuprofen). When prescribed with anticoagulant and thrombolytic drugs (alteplase, streptokinase, urokinase), the risk of bleeding simultaneously increases. Cefamandole, cefaperazone, cefotetan, valproic acid, plicamycin increase the incidence of hypoprothrombinemia. Myelotoxic drugs increase the manifestations of hematotoxicity of the drug. Cyclosporine and Au preparations enhance the effect of ibuprofen on Pg synthesis in the kidneys, which is manifested by increased nephrotoxicity. Ibuprofen increases the plasma concentration of cyclosporine and the likelihood of developing its hepatotoxic effects. Drugs that block tubular secretion reduce excretion and increase plasma concentrations of ibuprofen.
Dispensed from pharmacies: Dispensed by prescription.
Drug registration number: P No. 015343/02
Date of registration (re-registration) of the drug: December 30, 2003
The effectiveness of the combination Nurofen forte + gel in the treatment of osteochondrosis
About the article
1595
0
Category: General articles
Author: Sharov M.N. 1 1 FSBEI HE MGMSU im. A. I. Evdokimov, Ministry of Health of Russia, Moscow; State Clinical Hospital named after. S.I. Spasokukotsky DZM, Moscow
Spinal osteochondrosis is one of the most common conditions in general medical practice. This disease occupies one of the leading places among the reasons for seeking medical attention and disability in people of all ages. Spinal osteochondrosis is one of the well-studied causes of vertebrogenic dorsalgia. The process is primarily localized in the nucleus pulposus of the intervertebral disc, which becomes less elastic due to loss of moisture. Under the influence of mechanical stress, the nucleus pulposus can sequester and protrude towards the annulus fibrosus of the disc. Over time, cracks form in the annulus fibrosus. The development of spinal osteochondrosis and its progression are caused by congenital bone anomalies, excessive physical activity and other reasons that contribute to the wear and tear of cartilage tissue. The reasons for the development of osteochondrosis are not clear enough. Most likely, age-related wear and tear plays an obvious role. However, the first signs of degenerative changes in the spine are detected already at a young age. The source of pain in compression syndromes is pathologically altered structures of the spinal column, which either irritate tissue nociceptors or compress the spinal roots. In reflex syndromes, the source of pain can be both the spine itself and reflexively spasmed muscles that form tunnel syndromes. Drug treatment should be structured taking into account all links in the pathogenesis of pain and include impact both on the lesion in the spinal motion segment and on factors contributing to the appearance of pain.
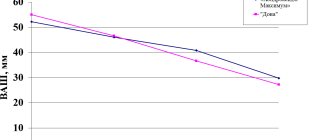
In the acute period of the disease, when the pain syndrome is severe, the main task of the doctor is to relieve pain. To successfully complete this task, certain conditions must be met, one of which is the choice of drug. The use of non-steroidal anti-inflammatory drugs (NSAIDs) is pathogenetically justified, since in addition to the analgesic effect they also have an anti-inflammatory effect. A feature of modern NSAIDs is the variety of dosage forms, including those for topical use in the form of ointments, gels, sprays, as well as suppositories and preparations for parenteral administration. The main key mechanisms are universal for most drugs, although their different chemical structures suggest a predominant effect on some specific processes. Currently, along with many other representatives of this group of analgesics, the drug Nurofen forte, which has not only a tablet form, but is also available in the form of a gel, has become widely recognized. Moreover, the clinical effectiveness of this analgesic is confirmed not only when used as a drug of choice for osteochondrosis, but also for other syndromes accompanied by pain. Pharmacodynamics of the drug Together with acetylsalicylic acid and diclofenac, Nurofen belongs to the acid antipyretic and anti-inflammatory painkillers. Its analgesic, antipyretic and anti-inflammatory effect is 2.5–3 times stronger than that of acetylsalicylic acid. The mechanism of its action is reversible competitive inhibition of cyclooxygenase, and Nurofen forte competes with arachidonic acid when binding to the active center of the enzyme. Inhibition of cyclooxygenase prevents prostaglandin synthesis and therefore prevents sensitization of pain receptors through prostaglandin E2 or I2. In addition to this mainly peripheral action of ibuprofen, it also has an analgesic effect in the central nervous system in the apical horn of the spinal cord and in the higher structures of the pain pathways. In therapeutic doses, Nurofen forte reduces the formation of bradykinin and the release of biogenic amines, which play a significant role in the inflammatory process, by 70–80%. The analgesic activity of the combination of tableted Nurofen forte in combination with the gel form of the drug in relation to inflamed tissues is not inferior to the activity of narcotic analgesics, in contrast to which NSAIDs do not affect the ability of the central nervous system to summarize subthreshold irritations. The analgesic effect of NSAIDs in osteochondrosis is more pronounced with pain of mild and moderate intensity, which is localized in the muscles, joints, tendons, and nerve trunks. For severe vertebrogenic syndromes in combination with degenerative processes such as osteochondrosis, the combination of Nurofen forte + gel is no less effective than potent analgesics. The tablet form of Nurofen is well absorbed in the gastrointestinal tract, almost completely binds to plasma albumin (99%), and displaces some other drugs. Maximum plasma concentrations are achieved within 1–3 hours. Metabolized in the liver into inactive metabolites, 90% of the dose is excreted by the kidneys, partly in free form, partly in the form of conjugates. Small amounts are excreted in bile. The half-life is approximately 2 hours. T1/2 for the tablet form (slow release of ibuprofen) – 10–12 hours. Complete elimination is completed after 24 hours. Compared to other non-steroidal painkillers, Nurofen has relatively few side effects, which is the reason for its unusually frequent use. It occupies a leading position among the non-steroidal painkillers most commonly used by doctors. Pharmacokinetics of Nurofen forte The pharmacokinetics of Nurofen have been well studied. In terms of physicochemical characteristics, Nurofen, as mentioned above, belongs to acid painkillers and consists of a hydrophilic and lipophilic part, which is important for their pharmacokinetic characteristics. Ibuprofen (as the active substance of Nurofen) is almost insoluble in water, but dissolves in dilute aqueous solutions of alkali hydroxides and carbonates; sodium salt (Na–Salz) is highly soluble in water. Ibuprofen is also highly soluble in various organic solvents. The pKa value for Nurofen is 4.5, for acetylsalicylic acid - about 3.5 and for ketoprofen - 5.5. As the dosage increases, the proportion of the active substance that is not bound to plasma proteins increases. Acidic painkillers are absorbed in the upper gastrointestinal tract at different rates. Nurofen forte is mainly absorbed in the small intestine. Water solubility plays a critical role in absorption; for Nurofen it strongly depends on the pH value. At pH 2 it is the smallest and then increases very strongly; at pH above 7 Nurofen is almost freely soluble in water. On the other hand, lipid solubility is highest at pH 2 to 4 and decreases with increasing pH value. Taking 400 mg of Nurofen on an empty stomach results in a highest plasma concentration of approximately 34 mg/ml. Biological digestibility ranges from 80 to 100%. After metabolization in the liver, pharmacodynamically passive metabolites are excreted primarily through the kidneys. Absorption of Nurofen forte Nurofen forte is absorbed quickly after oral administration. Highest plasma concentrations are achieved after oral administration within 1–2 hours. It is suggested that the rapid absorption of the drug determines the short time of its contact with the gastric mucosa and, accordingly, a relatively low risk of ulcerogenic effects. It is quickly absorbed in the intestines and quickly eliminated, does not accumulate even when metabolic processes are disrupted. If a rapid onset of action is required to reduce pain, oral administration with the fastest onset of action of the active substance is recommended. Nurofen forte and treatment of osteochondrosis For the treatment of regional pain syndromes of the spine, accompanied by severe pain, the combined use of Nurofen forte in the form of gel and tablets is recommended. Combination Nurofen forte at a dose of 400 mg/day. and its transdermal form in the form of a gel for spinal osteochondrosis has powerful analgesic and anti-inflammatory activity, combined with good tolerability, and therefore clinical effectiveness. Nurofen gel, which is applied directly to the painful area, is quickly absorbed and has a good analgesic effect, especially at the onset of the disease. The peripheral antinociceptive effect may result from the activation of certain types of K+ channels in the neuron membrane, causing hyperpolarization of the peripheral terminals of primary afferents. An important feature of the pharmacological action of Nurofen forte is the absence of a negative effect on the metabolism of cartilage tissue. The widely used extended-release form of Nurofen contains 400 mg of active substance. Among NSAIDs used topically, Nurofen gel can be distinguished, which meets safety requirements and has a number of advantages. The gel form of the drug ensures rapid penetration into deep tissues than when using other forms (ointments, creams), and causes an additional analgesic effect. The active ingredient of the drug is ibuprofen (5%). A number of studies have shown that ibuprofen has an affinity for connective tissue. Conclusions The biological absorption of Nurofen forte after oral administration is determined primarily by its solubility, its release from the dosage form and its absorption from the gastrointestinal tract. Absorption of Nurofen forte occurs quickly and completely; following oral administration, highest plasma levels are achieved within 1–2 hours. The solubility of a substance with the physicochemical characteristics of ibuprofen in water is the main condition for its absorption in the small intestine. This limits the absorption of Nurofen forte through the rate of its release from the corresponding drug. Since painkillers often require a rapid onset of action, fast-release products offer a therapeutic advantage. In the structure of the prevalence of chronic pain syndromes, musculoskeletal pain takes first place. To prevent the development and chronicity of pain, it is necessary to start symptomatic analgesic therapy with NSAIDs (for example, with the specified combination) as early as possible. It should be remembered that NSAIDs are especially effective in the early stages of pain development. It is at this early stage - the stage of acute and subacute pain - that with the help of traditional NSAIDs (Nurofen, etc.) it is possible to influence the peripheral components of the pain syndrome.
Content is licensed under a Creative Commons Attribution 4.0 International License.
Share the article on social networks
Recommend the article to your colleagues