Paget's disease is a dangerous pathology in which the restoration of bone tissue slows down, and an abnormal formation appears in its place. The disease can progress on any bones of the human skeleton and usually affects 3 or more bones. In the absence of timely diagnosis and prompt access to qualified medical help, treatment will be extremely difficult and will require a very long period of time.
Causes
Contents
hide
1 Causes of occurrence
2 Paget's disease: symptoms
3 Similarities with osteoarthritis 3.1 Paget’s disease: diagnostic methods 3.1.1 Treatment methods for Paget’s disease
To this day, the cause of this disease is not known to modern medicine. At the same time, there is a theory that it arises due to the presence of a genetic predisposition in the patient. Also, a number of sources mention that this disease can form due to the presence of improper functioning of phosphorus and calcium metabolism in the body. The third cause of the disease is viral infections, such as measles. Most often, strong representatives of humanity over 40 years of age suffer from this disease. If we talk about children, it should be noted that this disease practically does not occur in them.
1.General information
Paget's disease is a rare independent form of breast cancer with localization of the lesion in the nipple or areola. This disease was first described by the famous anatomist and surgeon A. Velpeau (France, 1856), but named after the English surgeon James Paget, who studied this type of cancer in detail (1874), emphasizing its pathogenetic connection with the subsequent breast carcinoma.
It should be noted that two more oncological diseases are named after J. Paget - neoplasia of bone and female genitalia - therefore, in each case it is necessary to clarify which Paget's disease we are talking about.
Epidemiological estimates range from 0.5-5% of total breast cancer. In the vast majority of cases (over 95%), other breast neoplasias are also diagnosed with Paget's disease. Both women and men get sick (usually at a later age than women and in a more aggressive form). The average age at which Paget's disease is diagnosed is about 65 years; In young people, this form of cancer is sporadic.
A must read! Help with treatment and hospitalization!
Paget's disease: symptoms
Today, medicine notes a direct relationship between symptoms, the severity of the disease, and bone damage. The bones most often affected by this disease are the bones of the legs and arms, skull, pelvis and spine. If we talk about the symptoms of the formation and development of the disease, they can occur in different ways:
In some cases, the disease occurs without any symptoms.
The disease has a high severity, in which there is severe pain and bone deformation occurs.
However, most often medicine notes the presence of an intermediate variant of the course of the disease in patients. This option has the following characteristics:
- Painful sensations during palpation.
- The presence of thickened areas on the bones that are easy to detect by simple palpation.
- The presence of a dull aching pain in the area of the affected bone.
Osteitis deformans: 100 years after J. Paget
IN
1999 marked 100 years since the death of the English surgeon and pathologist James Paget (born in 1814), whose name in medicine is associated with 3 diseases:
osteitis deformans
; other names: Paget's bone disease, deforming ostosis, deforming osteopathy), as well as
Paget's cancer
(eczema-like breast cancer) and
Paget-Schrötter syndrome
(acute subclavian vein thrombosis). In 1876 (20 years before the discovery of X-rays), J. Paget reported (published an article in 1877) about 5 patients with widespread skeletal lesions, mainly the skull and long bones of the lower extremities, accompanied by their thickening, softening and deformation, and He called this disease deforming osteitis, suggesting that it is based on inflammation of bone tissue. By 1882, he had already observed 23 cases of the disease and described in detail not only the clinical picture, but also sectional data.
The inflammatory nature of the disease was rejected in the 20s of our century, after which Paget’s bone disease (PDB) was for some time classified as a type of fibrous osteodystrophy, which also included fibrous osteitis, a systemic lesion of bone tissue described by the German pathologist F. Recklinghausen in 1891. , and fibrous bone dysplasia. But already in the 30s, each of these diseases was recognized as independent. With fibrous osteitis, hyperfunction of the parathyroid glands was established (A.V. Rusakov was one of the first to identify this connection in 1925), and fibrous bone dysplasia was classified as a congenital anomaly (the assumption of a defect in the development of osteoblasts in this disease was first made by V. R. Braitsev). KBP was isolated after the classical works of G. Schmorl (1930–1932). In our country, the pathomorphology of KBP was first described in detail by A.I. Abrikosov (1926).
Paget's disease of bone is an acquired chronic disease caused by local disorders of bone tissue remodeling.
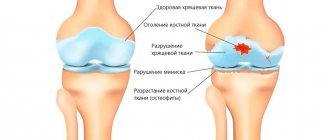
(see diagram). It is believed that the pathological process is initiated by increased resorption carried out by osteoclasts, which leads to a compensatory increase in the formation of new bone tissue. In this case, an excess amount of collagen fibers is synthesized, located in different directions, not in an orderly manner, which leads to the formation not of lamellar, as is normal, but of coarse-fibrous, excessively vascularized bone tissue, which naturally forms in the embryonic period, and in adulthood is observed only in places of healing of fractures and with hyperparathyroidism. Repeated processes of resorption and neoplasm lead to disorganization of the architectonics of bone tissue. The structure of the bone in the lesions of KBP resembles a mosaic, where zones of abnormal, newly formed fibrous bone tissue, foci of lamellar bone and areas of resorption are randomly located. A mosaic structure is often observed over a significant extent of the affected bone.
A characteristic feature of this disease is the locality of skeletal damage:
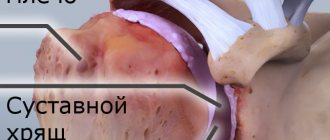
changes are noted in any one bone (monostotic form), in several bones (usually asymmetrical) or in many parts of the skeleton. The predominant localizations of “Paget's lesions” are (in descending order of frequency): the spine (usually the lumbar region), the skull (almost always the brain), the pelvis and the long tubular bones of the extremities (most often the femur, tibia and humerus). Less commonly, changes can be detected in the bones of the upper extremities, clavicles, shoulder blades, ribs, facial skull, hands and feet, and sometimes even in the sesamoid bones. Within one bone there may be several affected areas. Their slow growth is typical (no more than a few millimeters per year), but there are also cases of more intense progression. The number of lesions, by all accounts, does not increase over time. The pathological process, as a rule, does not extend to the joints. The exception is the sacroiliac joints, which can be completely destroyed by a layer of newly formed structureless bone tissue; There are also cases of transition of changes from the vertebral body to the intervertebral disc.
Prevalence
With targeted pathological and radiological examination, KBP is often detected, and in patients over 55–60 years of age, the chance of detecting this pathology clearly increases. In a non-selective X-ray examination of the skeleton in 4603 deceased, G. Schmorl established changes typical for KBP in 138 (3%) cases, and DH Collins noted that the frequency of osteitis deformans in old age (after 80 years) reached 10%. There are many descriptions of individual cases of KPD in middle and even young age, but early onset of the disease is still considered atypical.
Geographical differences in the prevalence of the disease are known. The incidence of CKD is highest in England (4.6%), Australia and New Zealand (3–4%), France (2.4%), slightly lower in Ireland (0.7–1.7%), Spain and Germany (1.3%) and significantly lower in Italy, Greece (0.5%) and especially in Norway and Sweden (0.3%). KBP is extremely rare in Central Africa (0.01-0.02%) and almost unknown in Asian countries.
In the UK, when analyzing a large array of skeletal X-rays in 1993–1995. There was a more than 2-fold decrease in the incidence of KBP compared to a similar study conducted in 1974.
In our country, the prevalence of KBP has not been studied, but it is known that at one time individual radiologists had a significant number of observations (600 cases or more).
Etiology
In 1974, during an ultrastructural study of osteoclasts obtained from the zones of “Paget's foci,” virus-like inclusions similar to the nucleocapsid of paramyxoviruses were identified in the nuclei and cytoplasm, and later it was established that they contained antigens of the nucleocapsid proteins of measles viruses and respiratory syncytial virus. In the 1980s, measles virus mRNA sequences were isolated from osteoclasts and other bone cells in patients with osteitis deformans. Measles virus transcripts have been shown to be present not only in Paget bone osteoclasts, but also in hematopoietic stem cells, more differentiated osteoclast precursor cells, and even in circulating peripheral mononuclear cells. A number of other studies have found evidence of canine distemper virus, also a member of the paramyxovirus class, and have found that the risk of developing CDV increases with mongrel dogs kept at home, especially if the animals have not been vaccinated against canine distemper virus.
It should be noted that attempts to identify signs of viral infection in patients with osteitis deformans were not always successful, and viral inclusions reminiscent of those found in KPD are observed in bone tissue cells with giant cell tumor, pycnodysostosis, osteopetrosis and primary oxalosis. It has not yet been possible to isolate the virus from KBP.
Hereditary predisposition
The possibility of the existence of CBP in close relatives has long been known. Using isotope scanning of the skeleton, it was shown that asymptomatic changes in at least one of the first-degree relatives are detected very often
, in approximately 40% of those examined. Family genetic studies have revealed that the risk of developing CBP in first-degree relatives is increased 7-fold.
The search for susceptibility genes was initially limited to the study of the histocompatibility system. In general, these attempts were not successful, although there are reports of a slight increase in the detection of certain HLA system antigens (DQw1, DR1, DR2, Drw6, DPw4) in KPD. In recent years, attention has been drawn to one of the loci (q21–22), located on chromosome 18. The reason for this was the establishment of a connection between this locus and a very rare bone dysplasia - familial common osteolysis, which has features similar to KBP. It was possible to show that there is an association with this locus in osteitis deformans. It has been suggested that CBP and familial common osteolysis may occur due to different mutations in the same gene or family of genes. The lack of a mandatory association with the 18q21–22 locus indicates, however, that genetic susceptibility to CBP is likely heterogeneous and there may be at least one more susceptibility locus to be discovered.
Clinical picture and diagnosis
The disease is diagnosed in the vast majority of cases after 40 years of age, most often around 60 years of age and very rarely before 25 years of age, with approximately the same frequency in men and women.
Two extreme variants of KBP can be distinguished: asymptomatic and clinically pronounced, characterized by persistent complaints and multiple bone deformations. Asymptomatic
the variant is noted in approximately 1/4 of all cases of the disease and, if detected during life, it is usually by chance, during the analysis of X-rays taken for another reason, or in the process of searching for explanations for elevated levels of alkaline phosphatase.
The generalized, polyostotic
variant of the pathology encountered by J. Paget is rare. In such patients, complaining of constant pain in the bones and large joints, remarkable skeletal deformities develop: pronounced kyphosis with shortening of the torso and O-shaped curvature of the lower extremities, leading to a “monkey” gait, as well as an increase in the size of the skull, which is typical for an elderly patient allows one to suspect the diagnosis of KBP at first glance. In the generalized version of the disease, the only systemic manifestation of KBP can be identified - heart failure, caused by an increase in cardiac output due to increased vascularization of altered bone tissue.
Usually there is an “intermediate”
a variant of KBP, which is characterized by local, predominantly nonspecific complaints and symptoms determined by the topic of the disease.
The disease can be suspected by complaints of pain in the bones (although the nocturnal nature of the pain is not typical), pain on palpation and detection of tissue hyperthermia over the affected area. Two other peculiar symptoms of KBP observed when the skull is affected are dilation and swelling of the veins in the vault and angioid streaks on the retina (a consequence of local intensification of bone blood flow). Complaints caused by secondary degenerative changes in the joints, spine, and skeletal deformities prevail. According to RD Altman and B. Collins, the most common complaint in 290 patients with osteitis deformans was pain in the lower back (in 34% of patients); secondary osteoarthritis of the hip joints was detected in 30%, knee joints – in 11% of patients; Deformities of the femur and tibia were found in 45% of patients.
With CBP, neurological disorders naturally develop, especially when the skull and spine are affected. Restructuring of the temporal bone (as well as changes in the auditory ossicles) leads to deafness; softening and deformation of the base of the skull may be accompanied by its flattening and basilar invagination of the spine, leading to compression of the brain stem, hydrocephalus and increased intracranial pressure. The growth of bone tissue in the area of the vertebral arches can cause compression of the spinal roots. Possible narrowing of the vertebral artery canal, accompanied by corresponding ischemic symptoms. Protrusion of an enlarged vertebral body into the spinal canal can lead to myelopathy, and damage to the sacrum can lead to the development of cauda equina syndrome.
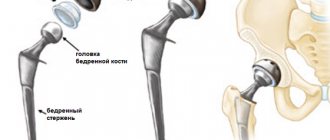
The incidence of long bone fractures, especially the femur and tibia, is increased in patients with KPD. Approximately 1/3 of cases of femoral fractures involve subtrochanteric or cervical fractures. Typical fractures may be preceded by so-called gap, incomplete fractures. In most cases, the fusion process occurs without deviations from the norm, within the usual time frame. When treating femoral neck fractures, hip replacement replacement is often necessary.
Another feature of KPD is the possibility of developing sarcomas (mainly osteosarcomas, but also fibrosarcomas and chondrosarcomas). Sarcomas occur in less than 1% of patients, developing in old “Paget's lesions” located in the bones of the pelvis, femur and humerus.
With KBP, it is natural, depending on the degree of activity and prevalence of the process, to increase the daily excretion of hydroxyproline in the urine and the activity of alkaline phosphatase (ALP) in the blood, reflecting, respectively, the intensity of resorption and formation of bone tissue. Patients with the highest values of alkaline phosphatase (they can exceed the normal level by 5–10 times), as a rule, have a polyostotic form of the disease with damage to the skull. Regular monitoring of these parameters allows us to evaluate the course of the disease and the effectiveness of therapy.
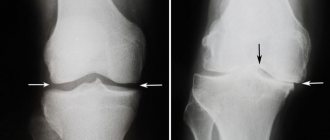
The diagnosis of KPD is made exclusively by radiography
. Characteristic changes in different parts of the skeleton are described exhaustively, including in the domestic literature. To clarify the degree of prevalence of KBP, skeletal scincigraphy is of approximate value.
Treatment
Over the past 20 years, radical changes have occurred in the treatment of CBP: symptomatic drugs have been replaced by drugs that can inhibit bone resorption
:
bisphosphonates, calcitonins and plicamycin.
These drugs are able to eliminate bone pain, tissue hyperthermia over the affected parts of the bones and, most importantly, slow down the excessive rate of bone tissue remodeling, as evidenced by a decrease in the levels of alkaline phosphatase in the blood, daily excretion of hydroxyproline, as well as normalization of the structure of bone tissue in the Paget lesion. With CBP, treatment is not required in all cases. Indications for the use of antiresorption agents are:
:
• the need to relieve pain (caused by the disease itself, and not its complications)
• polyostotic form of the disease
• mono- and oligoosseous forms of CBP in the case of prognostically unfavorable localization of the pathological process (near large joints, in the bones of the lower extremities, in the vertebral bodies, with extensive damage to the skull involving the temporal bone or base) and in the presence of disease activity (increased levels of alkaline phosphatase in the blood and/or hydroxyproline in daily urine)
• the need to prevent fractures
• carrying out orthopedic operations (in order to suppress the increased blood supply to the corresponding area of the bone and eliminate the increased risk of postoperative blood loss); This indication is not recognized by everyone.
Complications that have already arisen - bone deformations, deafness, other compressive neurological complications (radiculopathy, myelopathy) - are usually not amenable to this therapy.
If the size of the bone changes is small and there is no immediate threat of developing deformities or neurological complications, and there is no pain or biochemical signs of disease activity, a wait-and-see approach is followed.
The effect of antiresorption therapy is assessed by the dynamics of pain caused directly by CBP, but mainly by the degree of reduction in alkaline phosphatase activity. The analgesic effect is usually noted already in the first weeks, while the “biochemical effect” develops more slowly, its maximum severity is detected within 3–6 months.
Bisphosphonates
In recent years, preference has been given to bisphosphonates due to their selective effect on bone tissue, the ability to persist in it for a long time and the persistence of the effect caused. It should be noted that the pharmacology of bisphosphonates continues to actively develop. At least 3 generations of these drugs are now known (see table). The newest of them have 1000 times greater antiresorption activity compared to the first drug in this group, etidronate. Already during the creation of 2nd generation bisphosphonates (tiludronate, pamidronate and alendronate)
It was possible to expand the “therapeutic window” - the range between the therapeutic and toxic doses - it is too small for etidronate, which led to disturbances in the mineralization of newly formed bone tissue (osteomalacia) in a number of patients.
The effect of bisphosphonates in KPD depends on the severity of the disease, as well as on the dose..
It is selected (this especially applies to bisphosphonates used intravenously) individually, focusing on the prevalence of skeletal lesions, the prognostic significance of the localization of Paget's lesions, the nature of radiological changes (the predominance of signs of bone resorption indicates the need for more intensive therapy), and the degree of increase in biochemical markers of disease activity.
All bisphosphonates taken orally have a significant drawback - poor, unstable absorption (only about 1-10% of the dose taken) and a significant negative effect on it from a number of foods and even drinks. This forces us to recommend taking bisphosphonates 30 minutes before meals (usually before breakfast) and not consuming dairy products, calcium and iron supplements in the coming hours. To prevent “chemical” esophagitis, it is also recommended to drink the drug with at least 100 ml of water and remain in an upright position for about 30 minutes. This is particularly important with etidronate and alendronate and less so with the 3rd generation bisphosphonate residronate.
More successful results in the treatment of KBP are achieved with intravenous administration of bisphosphonates. Treatment is usually carried out in courses, the duration of each of them is from 3 to 6 months. For intravenous bisphosphonates, it is believed that the total dose rather than the duration of exposure is more important. By the 6th month of treatment, when the maximum “biochemical” effect of the chosen therapy regimen is usually revealed, a preliminary result is drawn. After this, it is customary to take a break from treatment, regularly (once a month) monitoring biochemical indicators of the rate of bone tissue remodeling. An indication for a repeat course is the resumption of bone pain and/or a new increase in alkaline phosphatase activity, exceeding the previously achieved level by 25%.
Some patients develop resistance to the drug over time, which must be overcome either by increasing the dose or by prescribing another bisphosphonate. There are patients in whom even with the help of new bisphosphonates it is not possible to achieve normalization of biochemical markers of disease activity, but in many of them the level of these indicators still decreases or decreases to a greater extent than with treatment with “old” drugs.
So far, no long-term studies have been conducted to prove a decrease in the incidence of long-term complications of KBP with long-term therapy with bisphosphonates, and the effect on the dynamics of radiological changes and the risk of developing sarcomas has not been clarified. However, there is hope for this, since repeated biopsies obtained after successful treatment have revealed normalization of the structure in the deposits of new bone tissue in the affected areas of the skeleton.
The main undesirable effect of bisphosphonates when administered orally is “irritation” of the upper gastrointestinal tract, including erosive esophagitis, and when administered intravenously, short-term fever accompanied by chills and myalgia (the so-called acute phase reaction, which is associated with an increase in interleukin levels 6 in the blood).
Etidronate
– the first, most tested drug for KBP and, importantly, the cheapest drug. The daily dose is selected per 1 kg of the patient’s body weight (5 mg/kg). Doses of etidronate as low as 10 mg/kg per day can cause osteomalacia (although they are more effective in inhibiting bone resorption), and doses less than 5 mg/kg are ineffective. In approximately a third of patients with osteitis deformans, no therapeutic effect is observed, which is largely due to poor absorption of etidronate. The effectiveness of etidronate may decrease during repeated courses of its use, and resistance not uncommonly develops. According to ES Siris (1998), etidronate (as well as calcitonins) is able to normalize impaired bone metabolism only in a small number of patients with KPD, mainly with a mild form of the disease.
The development of osteomalacia during treatment with etidronate can be manifested by pain and weakness in the proximal muscles, arthralgia, sometimes arthritis, usually of the ankle joints, enthesopathies in the heels, increased levels of alkaline phosphatase and (in rare cases) pathological bone fractures. Etidronate may occasionally cause alopecia, pancytopenia and hypersensitivity reactions (angioedema, rash, pruritus, Stevens-Johnson syndrome).
Clodronate
also belongs to the 1st generation bisphosphonates, but its effectiveness is higher than etidronate. It is used both orally and intravenously. When administered intravenously, the acute phase reaction characteristic of other bisphosphonates is usually not observed. Previous treatment with bisphosphonates may worsen the effect of clodronate.
Tiludronate.
A double-blind comparative study showed that tiludronate at a daily dose of 400 mg, used for 3 months, was significantly more effective than etidronate (400 mg per day), used for 6 months. A reduction in ALP levels of at least 50% after 6 months was achieved in 70% of patients treated with tiludronate, but only in 25% of patients treated with etidronate. Resistance was observed significantly more often in patients taking etidronate (in 51.9 and 19.5% of cases, respectively). The effect of treatment with tiludronate, unlike etidronate, was not dependent on previous use of bisphosphonates.
Pamidronate.
The effectiveness of pamidronate in KBP
is superior to previously created drugs
, especially in cases of relatively moderate activity or monoosseous form of the disease. In these cases, a single infusion of the drug (60–90 mg) often leads to long-term remission. There are known observations in which pamidronate was effective in patients previously unsuccessfully treated with etidronate. Normalization or almost normal levels of alkaline phosphatase can be achieved with a significant (5-10 times) initial increase in the level of this enzyme, but this requires the use of multiple infusions of the drug at weekly or 2-week intervals.
Pamidronate (as well as residualonate) in rare cases can lead to the development of anterior uveitis and scleritis. The pathogenesis of this side effect is unclear.
Alendronate.
Data on the use of alendronate in KBP are limited. Therapy for 6 months in patients with moderate or significant disease activity was accompanied by normalization of alkaline phosphatase levels in 48–63% of cases; when the treatment period was increased to 9 months, an additional effect was sometimes observed.
Resideronate
– one of the newest bisphosphonates. It is used orally; unlike other bisphosphonates, it can be taken after meals (no earlier than 2 hours later). In one study, a positive effect (a reduction in ALP levels by at least 50%) was observed in 90% of patients with moderate or significant levels of BCP activity; it did not decrease in the case of previously noted resistance to other bisphosphonates. Another study showed that the reduction in ALP levels achieved after the use of residronate was maintained in all patients up to 33 months. Side effects from the upper gastrointestinal tract were observed in 15% of patients, but did not force discontinuation of the drug; erosive esophagitis was detected endoscopically in only 1 patient.
Calcitonin preparations
Although the effectiveness of calcitonins in CBP is comparable to that of etidronate, they are currently used only for limited indications. This is due to the lack of aftereffect of calcitonin preparations (they do not accumulate in bone tissue) and the possibility of developing secondary resistance (according to review data, the frequency of this phenomenon ranges from 5–6 to 40%). Calcitonins are preferred
only in cases
where there are pronounced signs of osteolysis
, threatening the development of bone fractures, and also if it is necessary to obtain the fastest possible effect: in case of severe pain, the appearance of the first signs of neurological disorders, heart failure.
Plicamycin
Plicamycin (formerly mithramycin) belongs to the group of antitumor antibiotics. It is assumed that this drug has a selective and pronounced effect on osteoclasts. Characterized by a powerful and quickly onset effect
. The analgesic effect occurs within a few days. After a 10-day course of treatment, the level of alkaline phosphatase decreases by approximately 60%; in the next 1–2 months, the “biochemical effect” increases and persists from several months to several years. The severity of this effect depends on the dose and frequency of administration of the drug.
The general consensus is that plicamycin should be used as a reserve drug in KBP in the most severe, resistant cases, or when it is necessary to achieve an effect as quickly as possible (for example, with recent development of compression of the nerve trunks). This is due to the hepatotoxic and nephrotoxic effects of the drug, as well as the possibility of developing thrombocytopenia.
Plicamycin is used intravenously (in the form of long-term infusions) at a dose of 10–15–25 mcg/kg for no more than 10 days.
A bolus route of administration is known (25 mcg/kg every 2–3 weeks). Literature:
1. Abrikosov A.I. On the pathological anatomy of common fibrous osteitis (ostitis fibrosa s. deformans generalisata). Russian Clinic, 1926;(5): 343–74.
2. Braitsev V.R. Osteodystrophia fibrosa localisata. Proceedings of the XIX Congress of Surgeons. L., 1927.
3. Kosinskaya N. S. Fibrous dystrophies and bone dysplasias. L., Medicine, 1973.
4. Lagunova I.G. Clinical and radiological diagnosis of skeletal dysplasia. M., Medicine, 1989.
5. Reinberg S.A. X-ray diagnosis of bone and joint diseases. M., Medgiz, 1955; 523–34.
6. Rusakov A.V. On the question of the histo- and pathogenesis of Recklingausen's ostitis fibrosa. In the book: Second All-Russian Congress of Pathologists. M., 1925.
7. Altman RD, Collins B. Musculoskeletal manifestations of Paget's disease of bone. Arthr. Rheum. 1980; 23: 1121–7.
8. Barry HC Orthopedic aspects of Paget's disease of bone. Arthr. Rheum. 1980; 23: 1128–30.
9. Basle MF, Fournier JC, Rozenblatt S. et al. Measles virus mRNA detected in Paget's disease bone tissue by in situ hybridization. J. Gen. Virol. 1986; 67:907–13.
10. Birch MA, Taylor W, Fraser WD et al. Absence of paramyxovirus RNA in cultures of pagetic bone cells and in pagetic bone. J. Bone Miner. Res.1994; 9:11–6.
11. Brown JP, Hosking DJ, Ste-Marie L.-G. et al. Residronate, a highly effective, short-term oral treatment for Paget's disease: a dose-response study. Calcif. Tissue Int. 1999; 64:93–9.
12. Collins DH Paget's disease of bone. Incidence and subclinical forms. Lancet 1956; 271:51.
13. Cooper C., Schafheutle K., Dennison E. et al. The epidemiology of Paget's disease in Britain: Is the prevalence decreasing? J. Bone Mineral. Res. 1999; 14: 192–7.
14. Fraser WD Pagetic disease of bone. Current Opinion in Rheumatology. 1997; 9: 347–54.
15. Gordon MT, Anderson DC, Sharpe PT Canine distemper virus localized in bone cells in patients with Paget's disease. Bone 1991; 12: 195–201.
16. Haslam SI, Van Hul W, Morales-Piga A, Balemans W et al. Paget's disease of bone: Evidence for a suspectability locus on chromosome 18q and for genetic heterogeneity. J. Bone Mineral. Res. 1998;13:911–7.
Similarities to osteoarthritis
Due to the fact that the symptoms of this disease resemble osteoarthritis, they are often confused. However, in reality there is a big difference between these two diseases. For example, the main difference between Paget's disease and osteoarthritis is as follows:
- The presence of severe pain, even if the patient is in a state of complete rest.
- Decreased motor ability of the joints.
- The presence of high rigidity in joints after they are at rest.
- The appearance of thickenings on the bones, which lead to their fragility and subsequent complete destruction.
- A person suffering from this disease has a significantly increased risk of fractures, even in the case of minor bruises.
- As a result of pathological processes occurring in the body, the chances of pinched nerve endings significantly increase.
Paget's disease: diagnostic methods
The main way to diagnose this disease is to perform an x-ray, as well as laboratory blood tests. It should be noted that it is a laboratory blood test that allows us to identify the degree and complexity of the disease. During the blood test, an alkaline phosphatase test is performed, which makes it possible to determine the level of progression of the disease. Also, alkaline phosphatase testing is necessary for all people who have relatives suffering from this disease.
Methods for treating Paget's disease
Despite its high complexity and severity, this disease is treatable. However, in order for treatment to proceed as quickly as possible, it is necessary to seek qualified help at the first sensation of pain in the bone area. In addition, when diagnosing Paget's disease, the patient needs to minimize the level of stress on the damaged bones. In some particularly severe cases of the disease, in order to avoid bone destruction and fractures, the patient is given splints on the damaged areas. In addition, in order to ensure the safety of bones, increase their level of strength, and also prevent their destruction, the patient is prescribed medications containing calcium. Also, in some cases, vitamin D may be recommended to the patient along with calcium. If treatment is carried out in the presence of severely damaged bones, then surgical intervention will be necessary. This type of surgery will result in an osteotomy or an artificial joint being installed. By performing systemic surgical interventions, it is possible to restore a part of the body deformed as a result of the disease.
2. Reasons
The etiopathogenesis of Paget's disease still needs clarification. Two main hypotheses dominate. According to the first, specific cancer Paget cells are products of a malignant mutation of the cells of the nipple itself. The second hypothesis is that cells with apoptosis turned off (the “clock mechanism” of cell death after a certain number of divisions) migrate into the nipple from the ducts, where by that time ductal carcinoma has already developed - thus, nipple cancer is a kind of atypical metastasis.
The risk factors are apparently the same as for cancer in general, but in this case constant trauma to the nipple and areola, long-term non-healing cracks, as well as a genetic factor also play a certain role.
Visit our Oncology page
3. Symptoms and diagnosis
The first manifestation of Paget's disease is usually a reddened area with a tendency to form scales. This area of the nipple or areola may be virtually asymptomatic or slightly irritated; since there is a high probability of spontaneous pseudoremission (the period when the oncological process continues to develop in a latent form), few patients pay attention to the changed lesion and, moreover, consult a doctor at this stage.
Subsequently, the disease takes on an eczema-like character and makes itself felt by burning sensations, painfully increased tactile sensitivity, itching, and pain. There are hormone-dependent Paget's tumors, in which noticeable discharge may appear from the nipple. Sometimes an eczema-like rash spreads from the nipple to surrounding areas. In approximately half of cases, mammological examination reveals lumps in the breast.
A preliminary diagnosis is established clinically and confirmed histologically; if the eczema-like lesions described above appear on the nipple or areola, a biopsy must be prescribed. Among the methods of additional instrumental diagnostics, the most informative are mammography, ultrasound and MRI.
About our clinic Chistye Prudy metro station Medintercom page!
X-ray changes.
X-ray data reflect the nature of the main pathological process and the phase of the disease prevailing at the time of the study. The most commonly affected bones are the pelvis, followed by the femurs, skull, tibia, lumbosacral and thoracic spine, collarbones and ribs (in that order). Small bones are affected less frequently. The lytic phase of the disease is not always detected unless the skull bones are affected, as in limited osteoporosis, with the appearance of sharply defined radiolucent areas in the frontal, parietal and occipital bones. In long bones, areas of lysis are usually detected first at one end, from where they spread to the other in the form of a growing V-shaped edge. Damage may be accompanied by cortical proliferation and other signs of malignant growth. Typically, the area of lysis is followed by a zone of increased density, reflecting the process of new bone formation in the mixed phase of the disease. In general, the bone appears enlarged and denser with an unevenly expanded, rough and striated cortex; sometimes these changes are focal. Transverse radiolucent lines (cortical fractures) are often observed on the convex side of curved long bones, especially the femur and tibia. Complete fractures may also occur, some of which begin in areas of cortical fractures. Reconstruction of the affected bone is usually carried out in the direction of pulling muscle force or gravity, which determines the characteristic lateral bends of the femurs or the anterior bend of the tibia, as well as the deposition of the bulk of dense bone on the convex side of the bends. At the mixed stage of skull damage, there is an increase and thickening of the outer bone plates with uneven areas of increased density, often in the form of spots. Basilar invagination involving the base of the skull is common. Changes in the pelvic bones also reflect a combination of processes of bone resorption and new formation; the latter is often accompanied by a characteristic thickening of the pelvic arch. In the sclerotic phase of the disease, bone density may be uniformly increased; delineation is often absent. It is usually observed in the facial bones, but sometimes in the vertebrae, to which the homogeneity and density give an "ivory" appearance similar to that of typical Hodgkin's disease, although in the latter case the affected vertebrae are not enlarged.
What is Paget's disease and why does it develop?
- It is more common in Western Europe, North America and Australia.
- Synonyms: osteitis deformans, osteodystrophy deformans, osteosis deformans
- Paget's disease (osteitis deformans) is a chronic disease characterized by increased bone resorption (destruction), leading to weakening, deformation and pain in the bones.
- Frequency: 3% of people over 40 years of age
- 90% of patients diagnosed with ostosis deformans are over 40 years of age. Men are affected 1.5-2.1 times more often than women.
- The exact etiology of Paget's disease has not been established;
- the connection with infection of osteoclasts by paramyxoviruses and, as a result, acceleration of bone tissue destruction and remodeling is discussed.
Treatment of Paget's disease abroad in Israel, Germany and other countries
Treatment of deforming osteodystrophy is aimed at suppressing the activity of cells that destroy bone tissue, eliminating pain, preventing complications and malignant degeneration.
Symptomatic therapy
Aimed at eliminating pain and eliminating the local inflammatory reaction. For this purpose, NSAIDs (paracetamol, ibuprofen, baralgin, sodium diclofenac, etc.), hormones, and blockades are used.
Pathogenetic therapy
To suppress the activity of the process, Israeli specialists prescribe medications that prevent the breakdown of bone tissue and promote full bone formation:
- Nitrogen-containing bisphosphonates
- Calcitonin
Surgical correction
The level of development of Israeli orthopedics allows not only to perform osteotomy for therapeutic purposes, but also to perform orthopedic plastic surgery for severe deformities to restore joint function or for cosmetic purposes. Performed daily:
- Osteotomy to correct deformity
- Surgical correction of fractures with fixation of bone parts with plates or pins
- Joint replacement
For minor deformities, surgery is performed endoscopically, which is much easier for the patient to tolerate and reduces recovery time.
If the spinal cord structures are compressed, neurosurgery may be required.
Symptoms of deforming osteodystrophy
In the initial stage, Paget's disease may be asymptomatic. Patients complain of frequent dull pain in the bones and back, which worsens with rest. As the disease progresses, bone deformation is noted. If joints are involved in the process, patients complain of stiffness of movement. Pathological fractures often occur with normal daily activities.
Sometimes deformed bones of the skull can put pressure on the optic, auditory, facial or trigeminal nerves, causing specific symptoms.
When the spine is damaged, several vertebrae are involved in the process, and myelopathy and cauda equina syndrome may occur.
Severe deformities of the vertebrae cause compression of blood vessels and nerves.