II. Treatment and monitoring
General provisions about treatment
Treatment of OI is symptomatic and depends on the severity of the course.
The goal of treatment is to reduce the incidence of fractures, help a “fragile” person become more mobile and independent, and reduce pain. In addition, it is important to promptly identify and monitor extraskeletal manifestations and help the patient get rid of the side effects of drug therapy. A patient with OI should be managed by a team of specialists consisting of a pediatrician, endocrinologist, physical therapist, orthopedic traumatologist, geneticist, dentist, audiologist, psychologist, social worker, etc. A multidisciplinary approach - a combination of drug treatment, surgery, orthopedics and rehabilitation - significantly improves treatment results.
There is currently no cure for OI. In addition, there is no drug therapy developed specifically for the treatment of this disease, neither for children nor for adults. Drug therapy that is currently being carried out is based on drugs developed to treat age-related osteoporosis or prevent bone loss in adults with cancer and used off-label - not for the intended purpose in the instructions.
Strengthening bones in an attempt to reduce the incidence and severity of fractures is a key issue in the treatment of frail children and adults. That is why treatment is aimed at reducing the impact of the symptoms of the disease on the human body and preventing complications in the future.
An interdisciplinary management plan is often required, which may include:
- Visits to a geneticist (consultations and DNA testing);
- Orthopedic treatment of fractures and deformities;
- Hearing test;
- Examination for dental problems;
- Screening for the presence of cardiovascular diseases (ultrasound of the heart);
- Screening for respiratory problems (lung test);
- Physical therapy;
- Occupational therapy with adaptation of devices and environment;
- Psychological assistance;
- Dietary advice - nutrition consultation;
- Social assistance.
For people with OI, nutritious nutrition is very important to maintain vitamin D and calcium levels at the required level and is beneficial for bone health. It is also important to monitor your diet because people with limited mobility are prone to weight gain, and excess weight puts extra stress on the bones.
Attention should be paid to the prevention of osteoporosis. For people with OI, the additional effects of osteoporosis on the skeleton may have more serious consequences than for others. Please note that some common medications on the list of possible side effects have a risk of developing osteoporosis.
Drug treatment
Several types of bisphosphonates have been used as treatment for children. There are international protocols for two drugs—pamidronic acid and zoledronic acid.
The effectiveness of the drugs is the same, the difference is the duration of administration:
- pamidronic acid: three days, three hours a day, once every two to three months;
- Zoledronic acid: one day for an hour, every three months.
Some clinics also use other bisphosphonates, for example, drugs based on ibandronic acid. Treatment can be obtained in public and private clinics.
Physical rehabilitation
Rehabilitation is the most important component of an integrated approach to the treatment of OI. Rehabilitation must begin at an early age to help the child adapt to environmental conditions and overcome the fear of fractures when learning new motor skills.
For people with OI, rehabilitation is the most important stage in recovery from injuries, fractures, operations, as well as in the prevention of recurrent fractures and deformities. And if we talk about newborns, it is aimed primarily at teaching parents how to safely handle a “fragile” baby, which is necessary to prevent complications and to help the child develop motor skills. It has been noted that in patients with OI, the shape of the head may be triangular and flattened, the sutures between the bones of the skull may be unfused and open, and the fontanel may be very large. This will change over time. Often there are also disorders in the hip joints, which are caused by the use of soft surfaces for laying the child, which severely limit his movement. For prevention, you need to frequently change the baby's position. When a baby lies on his stomach, the upper limbs and cervical spine straighten, and the hip flexors stretch. This helps you learn to roll over and continue sitting.
The very first steps in physical therapy are controlling the position of the head, strengthening the muscles of the neck, the muscles responsible for the position of the body in space and the sitting position with support on the legs. Physical activity is very important for “fragile” children, and exercise should begin as early as possible. You can start developing motor skills and active swimming in the bathtub from birth; it is better to start going to the pool as soon as the opportunity arises. At older ages, the focus is on increasing muscle strength, functional development, and developing independence.
Consultation with physical therapists and answers to questions about physical activity can be obtained from the Fragile People Foundation’s free project “Mobile Rehabilitation Service.” You can sign up for an online consultation with physical therapist Nadezhda Epishina by email. Address
Clinics where the “fragile” are treated
A complete list of Russian clinics that diagnose and treat osteogenesis imperfecta is presented here.
Are there any prospects?
We, the doctors of the Center for Congenital Pathology GMS Clinic, would really like to see osteogenesis imperfecta treated in a modern and effective manner throughout the country. With the current trend, we will soon have more patients than we can properly treat, and we will have to expand our staff again. We would like to train specialists from other cities and transfer some of our patients to them for treatment. But, unfortunately, today our Center has no alternative in this direction. The few other institutions that try to deal with this disease are severely deficient in one aspect or another, and sending patients there for the time being would be to their detriment. We constantly receive patients who received care in government institutions, and we have to correct other people’s mistakes and regret the lost time and missed opportunities.
If you know doctors who are ready to adopt our experience, study from foreign literature, who are not indifferent to the global result of their treatment, who are ready to introduce new methods of therapy, surgery, rehabilitation, feel free to give them our contact information. We want there to be more centers like ours. Patients fly to us from the Far East and Yakutia. It would be much easier if they did not have to spend money on moving across the country, but rather travel, for example, to Novosibirsk or Yekaterinburg. But there are no alternatives to our center, even in Moscow.
If you know people or organizations that are ready to pay for our patients’ travel, technical equipment or accommodation in Moscow while they undergo a rehabilitation course at our Center (funds pay for treatment, but not rehabilitation courses after fractures or operations), share information about us with them. We will help them provide targeted assistance; we have lists of children who need this help more than others.
If you know children with brittle bones whose parents may not even know that the disease can be successfully treated, tell them about us. We will be happy to invite them to a consultation and, if necessary, will take them into our treatment program.
III. Forecasts
Due to the enormous individual variability of the disease, it is impossible to make general statements about what treatment and rehabilitation methods will be correct in each individual case. Some predictions can be made based on statistics for each type of OI, but no guarantees can be made regarding the number of fractures, the height of a frail person, or his mobility.
Even among “fragile” people within the same family, differences can be quite significant, and in each specific case it is recommended to seek individual advice from a specialist.
In many cases, bone fragility decreases after puberty, but may increase again after age forty, so physical activity is important throughout the life of a person with OI.
Classification of osteogenesis imperfecta
Type I: Slight bone fragility, sclera color: blue, dental changes: only in type IB, hearing loss: in most patients, inheritance type: A-D
Type II : Extreme bone fragility, sclera color: blue, dental changes: sometimes, hearing loss: no data, inheritance type: C, less often A-P
Type III : Severe bone fragility, sclera color: bluish at birth, dental changes: sometimes, hearing loss: often, inheritance pattern: A-P or A-D
Type IV : Varying degrees of bone fragility, sclera color: blue, dental changes: only in type IVB, hearing loss: common, inheritance type: A-D
Osteogenesis imperfecta type I is the mildest and is inherited in an autosomal recessive manner. Most patients have distinctly blue sclera.
Osteogenesis imperfecta type I is divided into subtypes A and B (the latter is distinguished by the presence of dentinogenesis imperfecta).
With osteogenesis imperfecta type II, death of the fetus or newborn occurs. Based on the features of skeletal changes detected by x-ray, osteogenesis imperfecta type II is divided into 3 subtypes (A, B and C).
Osteogenesis imperfecta types III and IV are easier than type II; It differs from type I in that the sclera is usually of normal color and only at a difficult age can it become bluish. Osteogenesis imperfecta type III (as opposed to type IV) progresses with age.
In addition, type III is inherited in an autosomal dominant or autosomal recessive manner, while type IV is inherited only in an autosomal dominant manner.
The course can be different, the type of inheritance is not always easy to establish, since the disease is often caused by a new mutation, and many parents, after the birth of a child with a severe form of osteogenesis imperfecta, are wary of having other children. For ales and other reasons, it makes little sense to distinguish Type IV. It is enough to divide osteogenesis imperfecta mild (type I), fatal (type II) and moderate (type III).
Prevalence
The prevalence of osteogenesis imperfecta type I is 1 in 30,000, type II is approximately 1 in 60,000 births, and the prevalence of the three severe forms recognized at birth (types I, III, and GU) is as high as 1 in 20,000.
Bone changes
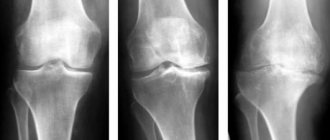
With osteogenesis imperfecta type I, bone fragility may lead to limitation of physical activity, or may not cause any discomfort to the patient. The cranial vault appears mottled on radiographs due to small areas of impaired ossification.
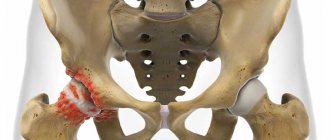
In osteogenesis imperfecta type II, the bones and other connective tissues are so weak that it results in severe injury to the fetus during pregnancy and childbirth (Fig. 348.5). The mineralization of many bones is not complete; multiple thickenings and fractures of the ribs, and deformation of the long tubular bones are characteristic.
For unknown reasons, long tubular bones may thicken or, conversely, thin. In osteogenesis imperfecta types III and IV, multiple fractures due to minor injuries cause gross deformation of the bones.
Kyphoscoliosis leads to respiratory failure, frequent pneumonia and the formation of cor pulmonale. An unfavorable sign is the appearance of areas of calcification at the ends of long tubular bones, resembling popcorn on an x-ray. Platybasia and communicating hydrocephalus can lead to progressive neurological disorders.
All forms of osteogenesis imperfecta are characterized by osteopenia. The degree of osteopenia can be difficult to assess because repeated fractures lead to limited physical activity and further decreased bone density. But, oddly enough, the fractures heal quite well.
Eye changes
The sclera may be white, slightly bluish, or bright blue. The blue color comes from the choroid of the eye, shining through a thinner-than-usual layer of collagen in the sclera. In some families, blue sclera is inherited, but is not accompanied by brittle bones.
Dentinogenesis imperfecta
Dental damage is not always observed; it may be significant or manifest as a mild discoloration. The enamel is usually unchanged; the characteristic amber, yellowish-brown or gray-blue color of the teeth is associated with dentin deficiency. Primary teeth are usually small, while permanent teeth may be bell-shaped (tapering towards the base). In some patients, teeth break easily and have to be removed.
Changes in dentin are due to the fact that it contains a lot of type I collagen. Similar dental changes sometimes occur in the absence of osteogenesis imperfecta.
Hearing loss
Hearing usually begins to decline between the ages of 10 and 20, and by the age of 30, hearing loss develops in 90% of patients. Hearing loss can be of varying degrees, conductive, sensorineural or mixed. Usually there are anomalies in the development of the middle ear, insufficient mineralization in some areas and pathological foci of calcification in other areas of the auditory ossicles.
Other manifestations
Damage to other organs and tissues is manifested by thin skin, a tendency to form scars, looseness and habitual dislocations of the joints, as with Ehlers-Danlos syndrome. Damage to the cardiovascular system can manifest itself as mitral valve prolapse, mitral and aortic insufficiency, and ruptures of large blood vessels.
In some patients, for unknown reasons, serum T4 concentrations and basal metabolism are increased, which is manifested by hyperthermia and increased sweating.
Molecular disorders
As already mentioned, most cases of osteogenesis imperfecta show mutations in one of the two genes encoding type I collagen.
In at least a third of patients, due to an unknown mutation, the concentration of mRNA of pro-α1(1) chains is reduced, which is synthesized less than pro-α2(1) chains. In severe forms of osteogenesis imperfecta (types II, III and IV), the effect of mutations is greatly enhanced as a result of the three mechanisms described above.
When mutations change the structure of the protein at the site of its cleavage by procollagen-M-endopeptidase, collagen accumulates with a retained N-terminal propeptide, which leads to joint laxity, as in Ehlers-Danlos syndrome type VII.
Mutations that change the structure of the middle or C-terminal parts of the molecule cause severe and often fatal forms of osteogenesis imperfecta. However, it is quite difficult to find a correspondence between a particular mutation and a specific clinical picture. Occasionally, homozygotes for mutant alleles encoding pro-α1 or pro-α2 chains are found.
Gonadal mosaicism
Most fatal forms of osteogenesis imperfecta are the result of new mutations with an autosomal dominant pattern of inheritance. However, the chance of having a second child with fatal osteogenesis imperfecta in the same family is about 7% due to gonadal mosaicism in one of the parents.
It has been found that in some men whose children suffer from osteogenesis imperfecta type II, some of the sperm carry the mutant allele. In addition, in healthy parents of such children, mosaicism of somatic cells, such as fibroblasts, leukocytes and hair root cells, can be detected. The possibility of gonadal mosaicism should be taken into account when providing medical and genetic counseling to healthy parents who have a child with a severe form of osteogenesis imperfecta.
Diagnostics
The basis for diagnosis is usually a combination of clinical picture (multiple fractures, blue sclera, dentinogenesis imperfecta) and family history. Other causes of pathological fractures should be excluded: child abuse, malnutrition, vitamin deficiencies, malignant neoplasms, as well as other hereditary diseases, such as chondrodysplasia or hypophosphatasia.
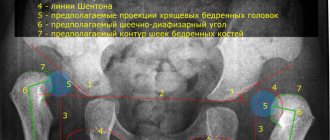
X-ray examination reveals bone loss, which can be confirmed using densitometry; The need for a bone biopsy is controversial. More than half of patients have a type I procollagen defect.
By incubating a culture of fibroblasts with labeled amino acids and subsequent electrophoresis of procollagen in a polyacrylamide gel, the following disorders that interfere with the assembly of triple helices can be identified:
1) decrease in the rate of synthesis of pro-a1(1) chains compared to pro-a2(1) chains;
2) shortening or lengthening of pro-a(1) chains;
3) changes in the structure of pro-a(1) chains as a result of amino acid substitutions and disruption of post-translational modification. The mutations themselves are detected by analyzing genomic DNA or cDNA.
Because each family usually has its own mutation, identifying it requires sequencing at least 5,000 bases in each of the two genes. If the mutation can be identified, prenatal diagnosis and identification of mutation carriers among the patient’s family members using PCR become possible.
Differential diagnosis for osteogenesis imperfecta
| Age | Diagnosis | Features |
| Newborns | Hypophosphatasia Achondrogenesis Thanatophoric dysplasia Asphic dysplasia of the chest Achondroplasia | Impaired mineralization of the skull bones Impaired mineralization of the vertebrae H-shaped vertebrae Narrow cylindrical chest Large head, short thick tubular bones |
| Chest | Child abuse Disturbance of osteogenesis with immobility of Scurvy Congenital syphilis | Typically traumatic brain injury and rib fractures |
| Children's | Homocystinuria Celiac disease Tumors of the adrenal cortex Treatment with glucocorticoids | Appearance as in Marfan syndrome, mental retardation Steatorrhea, anemia |
Causes of the disease and classification of pathology
The problem of bone fragility lies in the genetic predisposition of the body. Osteogenesis imperfecta
, pathology is associated with a violation of inheritance patterns. As a result of complex biochemical processes in the human body, gene mutations occur, which result in a lack of collagen synthesis, the component responsible for the formation of bone tissue. In its normal structure, bone tissue has a porous structure filled with loose connective tissue. As a result of the development of pathology, the cortical layer becomes thinner and the mechanical properties of the bones decrease. As a result of physical stress, patients develop pathological bone fragility. Like most genetic diseases, this pathology is considered incurable, but in recent years modern medicine has made significant progress in overcoming this disease.
Depending on the molecular and clinical changes, the pathology is divided into 4 types:
- the first type includes a mild form of pathology, in which brittle bones and the presence of blue sclera are observed;
- the second type includes perinatal-lethal pathology;
- the third type is associated with progressive skeletal deformation;
- the fourth type of pathology, dominant, is associated with unexpressed deformities.
Who pays for treatment
The drugs, like other treatment options, are available through compulsory medical insurance. But for this you need to undergo an examination and have a medical certificate.
Elena Meshcheryakova
You can receive medications prescribed from the federal center for free. They themselves are relatively inexpensive: one dropper costs about 30 thousand rubles. Considering that they are done to children two to four times a year, and to adults once every 12 months, this is quite affordable for the region’s budget.
But at the same time, there are quotas for treatment, which are not enough for everyone. Therefore, often medical care, such as orthopedics, is paid for by charities.
In our country, difficulties also arise due to the fact that medications are often prescribed not by trade name, but by active substance. Therefore, a generic equivalent of the main type of medication with less effectiveness can be issued at your place of residence. And here it is important that the child’s parents or the adult patient himself do not give up, but seek the right remedy.