Спондилоэпифизарная дисплазия
Как и большинство других форм врожденных остеохондропатий, спондилоэпифизарная дисплазия развивается по причине дефекта структурных белков костной и хрящевой тканей. Наиболее распространенная (частота 1:100 тыс.) аутосомно-доминантная разновидность заболевания обусловлена мутацией гена COL2A1, расположенного на 12 хромосоме. Он кодирует последовательность альфа-1 цепи коллагена 2 типа, наиболее распространенного в хрящевой ткани и стекловидном теле глаза. Помимо спондилоэпифизарной дисплазии дефекты этого гена приводят к развитию болезней Книста и Легга-Кальве-Пертеса, ахондрогенеза 2 типа и ряда других генетических патологий опорно-двигательной системы. Большинство нарушений в COL2A1 относятся к миссенс-мутациям, их результатом является экспрессия дефектного коллагена, неспособного полноценно выполнять свои функции. Для этой формы спондилоэпифизарной дисплазии наряду с поражениями скелета характерно развитие аномалий зрения – миопии, отслойки сетчатки, так как коллаген 2 типа представлен и в стекловидном теле.
Более редкая (встречаемость порядка 1:200-250 тыс.) разновидность спондилоэпифизарной дисплазии характеризуется рецессивным, сцепленным с Х-хромосомой наследованием. Причиной этой формы заболевания выступает мутация гена SEDL (TRAPPC2), кодирующего специфический белок седлин. По последним научным данным, этот протеин отвечает за ряд внутриклеточных транспортных процессов, в частности, способствует передаче белков от эндоплазматического ретикулума в комплекс Гольджи. Патогенез спондилоэпифизарной дисплазии при этом генетическом нарушении изучен недостаточно – предполагается, что дефект структуры седлина приводит к затрудненной модификации белков, в том числе входящих в состав костной и хрящевых тканей. Особенностью гена SEDL является полное отсутствие его ингибирования у женщин (что имеет место при некоторых сцепленных с полом заболеваниях), поэтому гетерозиготы являются только носителями патологического гена и у них не выявляется никаких нарушений. Эта форма спондилоэпифизарной дисплазии характеризуется более поздним началом, уменьшенной выраженностью симптомов и отсутствием поражения органов зрения или слуха.
Немаловажную роль в развитии симптомов спондилоэпифизарной дисплазии любого типа играет статическая нагрузка на опорно-двигательный аппарат. Так, при этом заболевании точки окостенения образуются в телах позвонков позже обычного – за этот период масса тела уже успевает деформировать костные структуры позвоночного столба, в результате чего окостенение происходит с аномалиями. Аналогичная ситуация и с эпифизами длинных трубчатых костей – именно поэтому при спондилоэпифизарной дисплазии преимущественно поражаются нижние конечности, испытывающие наибольшую нагрузку. Это обстоятельство используется в лечении данного заболевания – как можно более раннее применение ортопедических средств снижает нагрузку на элементы скелета, позволяя им сформироваться и окостенеть правильно. Такой подход значительно снижает выраженность костных деформаций при спондилоэпифизарной дисплазии.
Результаты
По данным клинического осмотра и УЗИ на первом и втором этапах наблюдения дисплазия ТБС диагностирована в 6855 (57,1%) случаях. Лечение в этих наблюдениях заключалось в разведении ног шиной Фрейка, курсе физиолечения и ЛФК. Диагноз был снят в 3072 (25,6%) наблюдениях с подтверждением результатами УЗИ в возрасте до 6 мес.
Особый интерес представляла группа из 5736 (47,8%) детей, которая рассматривалась нами как угрожаемая по развитию эпифизарной дисплазии. Группа включала 3783 (66%) ребенка в возрасте 6 мес с неразрешившейся дисплазией и 1953 (34%) ребенка с задержкой оссификации головок бедренных костей или многоядерным вариантом окостенения головок. Помимо специфических изменений костно-хрящевых суставных элементов у детей с дисплазиями ТБС выявлены множественные клинико-функциональные синдромы и фенотипические проявления недифференцированной дисплазии соединительной ткани (рис. 1).
Рис. 1. Фенотипические проявления дисплазии соединительной ткани.
Таким образом, у большинства детей с установленной дисплазией Майера имелась полиорганная патология диспластического типа.
Детям, имеющим к 6 мес жизни задержку формирования ядер окостенения головок бедренных костей, назначали консервативное лечение с включением традиционного восстановительного комплекса: общего расслабляющего массажа, электрофореза хлористого кальция на тазобедренные суставы с чередованием электрофореза трентала на поясницу, парафинотерапии на ТБС, лечебной гимнастики, а также ношение шины Виленского. Лечение проводилось в условиях физиоотделения курсами по 8–10 процедур. Кроме того, в лечебный комплекс входили медикаментозные средства, стимулирующие выработку коллагена (препараты магния, витамины группы В), препараты для стабилизации минерального обмена (водный раствор витамина D3, соли кальция), для нормализации уровня свободных аминокислот (глицин и др.), а также синхронизирующие биоэнергетический обмен (рибоксин, лецитин, милдронат и др.).
В результате лечения у 2954 (51,5%) детей нормальные размеры и структура головок бедренных костей восстанавливались к 9 мес, еще у 1618 (28,2%) — к возрасту 1 года. Однако у 1164 (20,3%) пациентов клинические проявления и объективные признаки дисплазии сохранялись и после 1 года. Эти дети нуждались в дальнейшем долечивании. В течение следующего года им проводили повторные курсы физиолечения и восстановительной медикаментозной терапии.
Прогностически значимым для реализации диспластических изменений ТБС в эпифизарную дисплазию считался возраст 2 года. Диагноз дисплазии Майера в этом возрасте окончательно установлен в 127 (2,2%) наблюдениях. В подобных случаях к курсовому лечению добавляли режимную разгрузку нижней конечности.
В результате описанной тактики ведения через 1–1,5 года диагноз дисплазии Майера снят в 100 наблюдениях, и лишь в 27 (0,4%) случаях дисплазия трансформировалась в асептический некроз головок бедренных костей.
Приводим клиническое наблюдение.
Ребенок А
., 1 год 10 мес, планово осмотрен ортопедом в связи с диспансерным наблюдением по поводу диагноза дисплазии Майера. Активных жалоб на момент осмотра не выявлено.
Из истории жизни: родился в 39–40 нед с функциональной незрелостью 36–37 нед. Масса при рождении 2700 г, рост 49 см. К возрасту 1 года состоял на диспансерном учете у невролога с диагнозом: синдром двигательных нарушений; у кардиолога с диагнозом: функционирующее овальное окно; у нефролога с диагнозом: пиелоэктазия справа; у хирурга с диагнозом: кавернозная гемангиома области спины, состояние после криодеструкции.
Из истории болезни: дисплазия ТБС выявлена в возрасте 2 мес при УЗИ. Лечение: разведение ТБС подушкой Фрейка (16 см), расслабляющий массаж (10 процедур), электрофорез 3% СаС12 на ТБС (10 процедур), ЛФК на разведение в ТБС. На контрольном УЗИ ТБС в 3,5 мес определялись признаки нормализации углов (α
— более 60°,
β
— менее 55°). Ядра окостенения отсутствуют. В возрасте 6 мес при УЗИ ТБС выявлены ядра окостенения слева 3 мм, справа — отсутствуют. Назначен повторный курс восстановительного лечения: массаж общий расслабляющий (10 процедур), электрофорез 1% трентала на поясницу (10 процедур), парафиновые аппликации на ТБС (8 процедур), кальций-компливит для малышей по 5 мл 1 раз в день 3 нед. В динамике в возрасте 9 мес при УЗИ ТБС определялось ядро окостенения слева 6 мм, справа — 3 ядра по 1 мм. Дополнительно в лечение добавлены: магне-В6 по 1,5 мл раствора для питья 2 раза в день, глицин по ¼ таблетки 2 раза в день. В возрасте 1 года 3 мес проведена рентгенография ТБС (рис. 2,
а
).
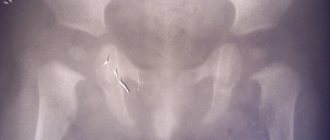
Рис. 2. Рентгенограммы тазобедренных суставов в прямой проекции больного А. а — в 1 год 3 мес; б — в 1 год 6 мес.
По данным рентгенографии, головки бедренных костей центрированы в вертлужных впадинах. Ацетабулярные углы слева — 21°, справа — 23°. Головка сплющена, фрагментирована, с элементами склерозирования. С учетом полученных данных рекомендована режимная разгрузка правой нижней конечности в течение 3 мес. Очередной курс массажа пояснично-крестцового отдела позвоночника из 10 процедур, электрофореза 3% СаС12 на ТБС, парафинотерапия на ТБС (по 8 процедур), милдронат 0,25 по ½ капсуле 1 раз в день 14 дней.
Объективно: ходит самостоятельно. Правильного телосложения. На коже множественные эктодермальные диспластические элементы: пигментные пятна, родинки. В области нижних ребер справа имеется рубец после криодеструкции. Мышечный тонус равномерно снижен. Увеличенный объем пассивных движений в мелких суставах кисти. Положительные симптомы «большого пальца», «запястья» (суставной счет по Beighton с модификацией для педиатрической практики 8 баллов). Позвоночник несколько отклонен от средней линии, небольшая асимметрия надплечий, треугольников талии. При наклоне вперед паравертебральной асимметрии нет. Ось таза наклонена. Относительное укорочение левой нижней конечности на 1,0 см, рекурвация в коленных суставах. Уплощение продольных сводов обеих стоп с вальгизацией пяточных костей при вертикальной нагрузке.
Локально: отведение бедер в полном объеме. Ограничена наружная ротация с двух сторон, больше справа. Увеличена внутренняя ротация бедер.
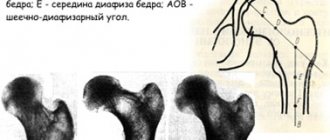
На контрольной рентгенографии (рис. 2, б
) ТБС в прямой проекции головки обеих бедренных костей центрированы в вертлужных впадинах. Шейка бедренной кости укорочена. Шеечно-диафизарный угол увеличен до 140°. Небольшое снижение высоты головки правой бедренной кости со значительным улучшением плотности головки.
По результатам динамических исследований пациенту А
. установлен клинический диагноз: дисплазия Майера, период восстановления; сопутствующий; синдром врожденной дисплазии соединительной ткани недифференцированный; вальгусная установка бедер; нарушение осанки, плоско-вальгусные стопы.
Даны рекомендации по рациональному двигательному режиму и пребыванию на диспансерном учете у ортопеда до 5-летнего возраста.
Публикации в СМИ
Эпифизарные дисплазии — наследственные заболевания, характеризующиеся нарушением эндохондрального окостенения; как правило, проявляются невысоким ростом, тугоподвижностью суставов, болями и деформациями конечностей, особенно нижних.
• Дисплазия эпифизарная, тип 1 Фэйрбэнка (#132400, 19pter-19qter, дефект гена олигомерного белка хрящевого матрикса СОМР [600310], Â). Клинически: врождённая карликовость с короткими конечностями, дисплазия бёдер, укорочение костей пястья и фаланг, брахидактилия, чрезмерный объём движений в суставах пальцев, незначительное расширение метафизов, укорочение шейки бедра, овальная форма тел позвонков, скошенный дистальный конец большеберцовой кости, задержка окостенения костей запястья.
• Дисплазия эпифизарная множественная, тип 2 (*600204, 1p33–p32, дефект гена COL9A2 [120260], Â). Клинически: начало в возрасте от 2,5 до 6 лет, боли в коленных и голеностопных суставах, деформация и увеличение коленных суставов, маленькие сглаженные эпифизы в большинстве суставов, особенно коленных.
• Дисплазия эпифизарная множественная, тип 3 (*600969, Â). Клинически: начало в детстве и пубертатном периоде, походка вперевалку, неподвижность и/или боли в коленных суставах, умеренная низкорослость, короткие руки, отсутствие патологии позвоночника, сглаженные эпифизы, деформация коленных суставов, постепенное развитие остеоартроза коленных и тазобедренных суставов.
• Дисплазия эпифизарная множественная Уолкотта–Раллисона с ранним развитием СД (*226980, r). Клинически: карликовость с коротким туловищем, СД 1 типа, множественная эпифизарная дисплазия, деминерализация кости, множественные переломы, ограничение отведения бедра, боли и тугоподвижность в суставах, изменение цвета зубов, спленомегалия, гепатомегалия, почечная недостаточность, спондилоэпифизарная дисплазия.
• Дисплазия эпифизарная множественная с миопией и кондуктивной тугоухостью (132450, Â). Клинически: прогрессирующая миопия, истончение сетчатки, катаракта, кондуктивная тугоухость.
• Дисплазия эпифизарная множественная (*226900, r). Клинически: множественная эпифизарная дисплазия, плоские головки бедренных костей, нормальные фаланги и кости пястья. Лабораторно: нитевидные или гранулярные включения в хондроцитах.
• Дисплазия семейная эпифизарная типа Бейкес (*142670, Â) — возможно, клинический вариант множественной эпифизарной дисплазии, названа по фамилии семьи с множеством поражённых в 6 поколениях. Клинически: боли в тазобедренных суставах, прогрессирующая скелетная дисплазия, выраженная сглаженность эпифизов головок бедренных костей, вторичный остеоартроз, кифосколиоз.
• Синдром Лоури–Вуда (*226960, r). Клинически: внутриутробная задержка развития, низкорослость, маленькие эпифизы, квадратные подвздошные кости, сглаженные вертлужные впадины, микроцефалия, нистагм, умеренная умственная отсталость.
• Дисплазия эпифизарная бедренной кости с задержкой роста и глухотой (601351, r). Клинически: задержка роста, лёгкая умственная отсталость, нейросенсорная тугоухость, двусторонняя атрезия слёзно-носовых протоков, паховая и пупочная грыжи, эпифизарная дисплазия бедра.
• Дисплазия макроэпифизарная с остеопорозом, складчатой кожей и поздним началом (248010, r). Клинически: низкорослость, недостаточное развитие подкожной клетчатки, сухие грубые волосы, складчатые ладони, аномалии ладонной дерматоглифики, снижение массы мышц, увеличенные эпифизы, остеопороз, повторные переломы.
• Дисплазия эпифизарная гемимелическая (болезнь Тревора, 127800; форма с остеохондроматозом, 127820). Клинически: асимметричное разрастание хряща эпифизов, особенно предплюсны и запястья. Преобладающий пол — мужской (3:1).
МКБ-10 • Q77.7 Спондилоэпифизарная дисплазия.