Spondyloepiphyseal dysplasia
Like most other forms of congenital osteochondropathy, spondyloepiphyseal dysplasia develops due to a defect in the structural proteins of bone and cartilage tissue. The most common (frequency 1:100 thousand) autosomal dominant type of the disease is caused by a mutation of the COL2A1 gene, located on chromosome 12. It encodes the alpha-1 chain sequence of type 2 collagen, which is most abundant in the cartilage and vitreous humor of the eye. In addition to spondyloepiphyseal dysplasia, defects in this gene lead to the development of Kniest and Legg-Calvé-Perthes diseases, type 2 achondrogenesis and a number of other genetic pathologies of the musculoskeletal system. Most disorders in COL2A1 are missense mutations, which result in the expression of defective collagen that is unable to fully perform its functions. This form of spondyloepiphyseal dysplasia, along with skeletal lesions, is characterized by the development of visual anomalies - myopia, retinal detachment, since type 2 collagen is also present in the vitreous body.
A rarer (occurrence rate is about 1:200-250 thousand) type of spondyloepiphyseal dysplasia is characterized by recessive, X-linked inheritance. The cause of this form of the disease is a mutation in the SEDL (TRAPPC2) gene, which encodes the specific sedlin protein. According to the latest scientific data, this protein is responsible for a number of intracellular transport processes, in particular, it promotes the transfer of proteins from the endoplasmic reticulum to the Golgi complex. The pathogenesis of spondyloepiphyseal dysplasia in this genetic disorder has not been sufficiently studied; it is assumed that a defect in the sellar structure leads to difficult modification of proteins, including those that make up bone and cartilaginous tissues. A feature of the SEDL gene is the complete absence of its inhibition in women (which occurs in some sex-linked diseases), therefore heterozygotes are only carriers of the pathological gene and no disorders are detected in them. This form of spondyloepiphyseal dysplasia is characterized by a later onset, reduced severity of symptoms, and the absence of damage to the organs of vision or hearing.
An important role in the development of symptoms of spondyloepiphyseal dysplasia of any type is played by static load on the musculoskeletal system. Thus, with this disease, ossification points form in the vertebral bodies later than usual - during this period the body weight already has time to deform the bone structures of the spinal column, as a result of which ossification occurs with anomalies. The situation is similar with the epiphyses of long tubular bones - this is why with spondyloepiphyseal dysplasia, the lower extremities, which experience the greatest load, are predominantly affected. This circumstance is used in the treatment of this disease - the earliest possible use of orthopedic means reduces the load on the skeletal elements, allowing them to form and ossify correctly. This approach significantly reduces the severity of bone deformities in spondyloepiphyseal dysplasia.
results
According to clinical examination and ultrasound at the first and second stages of observation, hip dysplasia was diagnosed in 6855 (57.1%) cases. Treatment in these observations consisted of spreading the legs with a Freik splint, a course of physiotherapy and exercise therapy. The diagnosis was removed in 3072 (25.6%) cases and confirmed by ultrasound results at the age of up to 6 months.
Of particular interest was a group of 5736 (47.8%) children, which we considered to be at risk for the development of epiphyseal dysplasia. The group included 3783 (66%) children aged 6 months with unresolved dysplasia and 1953 (34%) children with delayed ossification of the femoral heads or multinucleate ossification of the femoral heads. In addition to specific changes in the osteochondral articular elements, multiple clinical and functional syndromes and phenotypic manifestations of undifferentiated connective tissue dysplasia were identified in children with hip dysplasia (Fig. 1).
Rice. 1. Phenotypic manifestations of connective tissue dysplasia.
Thus, the majority of children with established Mayer's dysplasia had multiple organ pathology of the dysplastic type.
Children who had a delay in the formation of ossification nuclei of the femoral heads by the age of 6 months were prescribed conservative treatment including a traditional restorative complex: general relaxing massage, calcium chloride electrophoresis on the hip joints with alternating Trental electrophoresis on the lower back, paraffin therapy on the hip joint, therapeutic exercises, and wearing a Vilensky splint. Treatment was carried out in a physical department in courses of 8–10 procedures. In addition, the treatment complex included medications that stimulate collagen production (magnesium preparations, B vitamins), drugs to stabilize mineral metabolism (an aqueous solution of vitamin D3, calcium salts), to normalize the level of free amino acids (glycine, etc.), and also synchronizing bioenergy metabolism (riboxin, lecithin, mildronate, etc.).
As a result of treatment, in 2954 (51.5%) children, the normal size and structure of the femoral heads were restored by 9 months, and in another 1618 (28.2%) - by the age of 1 year. However, in 1164 (20.3%) patients, clinical manifestations and objective signs of dysplasia persisted after 1 year. These children required further follow-up treatment. Over the next year, they received repeated courses of physiotherapy and restorative drug therapy.
The age of 2 years was considered prognostically significant for the development of dysplastic changes in the hip joint into epiphyseal dysplasia. The diagnosis of Mayer's dysplasia at this age was finally established in 127 (2.2%) cases. In such cases, routine unloading of the lower limb was added to the course of treatment.
As a result of the described management tactics, after 1–1.5 years, the diagnosis of Mayer's dysplasia was removed in 100 cases, and only in 27 (0.4%) cases did dysplasia transform into aseptic necrosis of the femoral heads.
We present a clinical observation.
Child A
., 1 year 10 months, routinely examined by an orthopedist in connection with clinical observation regarding the diagnosis of Mayer's dysplasia. No active complaints were identified at the time of examination.
From the life history: born at 39–40 weeks with functional immaturity at 36–37 weeks. Birth weight 2700 g, height 49 cm. By the age of 1 year, he was registered with a neurologist with a diagnosis of movement disorders syndrome; from a cardiologist with a diagnosis of a functioning foramen ovale; a nephrologist with a diagnosis of pyeloectasia on the right; from a surgeon with a diagnosis of cavernous hemangioma of the back area, a condition after cryodestruction.
From the medical history: hip dysplasia was detected at the age of 2 months by ultrasound. Treatment: dilution of the hip joint with a Freik pillow (16 cm), relaxing massage (10 procedures), electrophoresis of 3% CaC12 on the hip joint (10 procedures), exercise therapy for dilution in the hip joint. At the control ultrasound of the hip joint at 3.5 months, signs of normalization of the angles were determined ( α
- more than 60°,
β
- less than 55°).
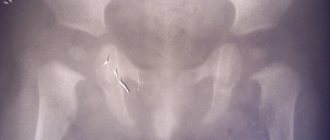
There are no ossification nuclei. At the age of 6 months, ultrasound of the hip joint revealed ossification nuclei of 3 mm on the left and no ossification nuclei on the right. A repeated course of rehabilitation treatment was prescribed: general relaxing massage (10 procedures), electrophoresis of 1% Trental on the lower back (10 procedures), paraffin applications on the hip joint (8 procedures), calcium-complivit for babies 5 ml once a day for 3 weeks. Over time, at the age of 9 months, ultrasound of the hip joint revealed an ossification nucleus of 6 mm on the left, and 3 nuclei of 1 mm on the right. Additionally added to the treatment: magne-B6 1.5 ml of drinking solution 2 times a day, glycine ¼ tablet 2 times a day. At the age of 1 year 3 months, radiography of the hip joint was performed (Fig. 2, a
).
Rice.
2. Radiographs of the hip joints in the direct projection of patient A. a - at 1 year 3 months; b - at 1 year 6 months. According to radiography, the heads of the femurs are centered in the acetabulum. The acetabular angles on the left are 21°, on the right - 23°. The head is flattened, fragmented, with elements of sclerosis. Taking into account the data obtained, routine unloading of the right lower limb was recommended for 3 months. Another course of massage of the lumbosacral spine of 10 procedures, electrophoresis of 3% CaC12 on the hip joint, paraffin therapy on the hip joint (8 procedures each), mildronate 0.25, ½ capsule once a day for 14 days.
Objectively: he walks independently. Correct physique. There are multiple ectodermal dysplastic elements on the skin: pigment spots, moles. In the area of the lower ribs on the right there is a scar after cryodestruction. Muscle tone is uniformly reduced. Increased range of passive movements in small joints of the hand. Positive symptoms of “thumb”, “wrist” (joint score according to Beighton with modification for pediatric practice 8 points). The spine is slightly deviated from the midline, slight asymmetry of the shoulder girdles and waist triangles. There is no paravertebral asymmetry when bending forward. The pelvic axis is tilted. Relative shortening of the left lower limb by 1.0 cm, recurvation in the knee joints. Flattening of the longitudinal arches of both feet with valgusation of the heel bones under vertical load.
Locally: full hip abduction. External rotation is limited on both sides, more so on the right. Increased internal rotation of the hips.
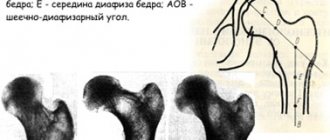
On control radiography (Fig. 2, b
) The hip joints in the direct projection of the heads of both femoral bones are centered in the acetabulum. The neck of the femur is shortened. The neck-shaft angle is increased to 140°. A slight decrease in the height of the right femoral head with a significant improvement in the density of the head.
According to the results of dynamic studies, patient A
. a clinical diagnosis was established: Mayer's dysplasia, recovery period; accompanying; undifferentiated congenital connective tissue dysplasia syndrome; valgus alignment of the hips; poor posture, flat valgus feet.
Recommendations are given on a rational motor regimen and staying at a dispensary with an orthopedist until the age of 5.
Publications in the media
Epiphyseal dysplasias are hereditary diseases characterized by impaired endochondral ossification; as a rule, they are manifested by short stature, stiffness of joints, pain and deformities of the extremities, especially the lower ones.
• Epiphyseal dysplasia, Fairbank type 1 (#132400, 19pter-19qter, defect in the cartilage matrix oligomeric protein gene COMP [600310], Â). Clinically: congenital dwarfism with short limbs, hip dysplasia, shortening of the metacarpal and phalangeal bones, brachydactyly, excessive range of motion in the finger joints, slight widening of the metaphyses, shortening of the femoral neck, oval shape of the vertebral bodies, beveled distal end of the tibia, delayed ossification of the carpal bones.
• Multiple epiphyseal dysplasia, type 2 (*600204, 1p33–p32, COL9A2 gene defect [120260], Â). Clinically: onset between the ages of 2.5 and 6 years, pain in the knee and ankle joints, deformation and enlargement of the knee joints, small flattened epiphyses in most joints, especially the knees.
• Multiple epiphyseal dysplasia, type 3 (*600969, Â). Clinically: onset in childhood and puberty, waddling gait, immobility and/or pain in the knee joints, moderate short stature, short arms, absence of spinal pathology, smoothed epiphyses, deformation of the knee joints, gradual development of osteoarthritis of the knee and hip joints.
• Wolcott-Rallison multiple epiphyseal dysplasia with early development of diabetes (*226980, r). Clinically: short-trunk dwarfism, type 1 diabetes, multiple epiphyseal dysplasia, bone demineralization, multiple fractures, limited hip abduction, joint pain and stiffness, tooth discoloration, splenomegaly, hepatomegaly, renal failure, spondyloepiphyseal dysplasia.
• Multiple epiphyseal dysplasia with myopia and conductive hearing loss (132450, Â). Clinically: progressive myopia, retinal thinning, cataracts, conductive hearing loss.
• Multiple epiphyseal dysplasia (*226900, r). Clinically: multiple epiphyseal dysplasia, flat femoral heads, normal phalanges and metacarpal bones. Laboratory: thread-like or granular inclusions in chondrocytes.
• Familial epiphyseal dysplasia type Bakes (*142670, Â) - possibly a clinical variant of multiple epiphyseal dysplasia, named after a family with many affected individuals in 6 generations. Clinically: pain in the hip joints, progressive skeletal dysplasia, pronounced smoothness of the epiphyses of the femoral heads, secondary osteoarthritis, kyphoscoliosis.
• Lowry-Wood syndrome (*226960, r). Clinically: intrauterine growth retardation, short stature, small epiphyses, quadrate iliac bones, smoothed acetabulum, microcephaly, nystagmus, moderate mental retardation.
• Epiphyseal dysplasia of the femur with growth retardation and deafness (601351, r). Clinically: growth retardation, mild mental retardation, sensorineural hearing loss, bilateral nasolacrimal duct atresia, inguinal and umbilical hernia, epiphyseal hip dysplasia.
• Macroepiphyseal dysplasia with osteoporosis, folded skin and late onset (248010, r). Clinically: short stature, insufficient development of subcutaneous tissue, dry coarse hair, folded palms, palmar dermatoglyphics abnormalities, decreased muscle mass, enlarged epiphyses, osteoporosis, recurrent fractures.
• Epiphyseal hemimelic dysplasia (Trevor's disease, 127800; form with osteochondromatosis, 127820). Clinically: asymmetrical growth of the cartilage of the epiphyses, especially the tarsus and carpus. The predominant gender is male (3:1).
ICD-10 • Q77.7 Spondyloepiphyseal dysplasia.